
Abstract
Determination of 226Ra and 228Ra radioisotopes in environmental samples and foodstuffs has been widely performed by using several radiochemical methods and radioactivity measurement techniques. A unique radiochemical method developed in our laboratory was applied to the measurement of 226Ra and 228Ra by using the same source with different measurement techniques. To validate the performance of this method, 226Ra and 228Ra in water and shrimp samples were measured by participating to IAEA-TEL-2019-03 Worldwide Proficiency Test and IAEA-TEL-2019-04 ALMERA Proficiency Test. The results were successfully passed in terms of both accuracy and precision criteria and obtained “Accepted” status.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among the many radium radioisotopes, 226Ra and 228Ra are important from the viewpoints of radiation protection and environmental protection due to their relatively long half-lives and plentiful presence in nature [1]. These radioisotopes are often present in industrial waste products stated as Technologically Enhanced Naturally Occurring Radioactive Material (TENORM). The consumption of foodstuffs and water having high radium activity concentration may lead to the accumulation of these radionuclides in human body and contribute to radiological dose. Therefore, the radium levels in industrial and consumer products should be controlled by the regulators for the human health and environment protection [2]. There are several methods including alpha-particle spectrometry, gamma-ray spectrometry, and liquid scintillation counting technique to measure the radium radioisotopes 226Ra and 228Ra [3, 4]. The alpha-emitter 226Ra can be measured directly by alpha-particle spectrometry, which is rated as one of the best methods for the environmental samples having extremely low 226Ra activity concentration in terms of accuracy and minimum detection limit (MDL) [4, 5].
The beta-emitter 228Ra can also be measured by alpha-particle spectrometry, but indirectly via its progeny 228Th. However, 228Ra measurement by alpha-particle spectrometry is not a practical way for the routine analyses since the long half-life of 228Th (1.9 a) causes long ingrowth periods, between 3 and 18 months depending on the 228Ra activity [2, 6, 7].
For the measurements of 226Ra and 228Ra radioisotopes, liquid scintillation counting (LSC) technique is one of the often used methods in environmental samples [8]. Although LSC has some drawbacks such as quenching, low energy resolution and relatively high background radiation, acceptable detection limits and high sample throughput put forward LSC as an adequate method for 226Ra and 228Ra radioisotopes [9,10,11,12].
Gamma-ray spectrometry is a non-destructive method permitting the simultaneous determination of 226Ra and 228Ra activities in a sample. However, large sample volume, relatively poor efficiency of the High Purity Germanium (HPGE) detectors, high background radiation and poor detection limits are the drawbacks of the gamma-ray spectrometry in measurements of environmental samples to attain accurate and reliable results. Nevertheless, gamma-ray counting technique is a good alternative and has been used by several laboratories [5]. 226Ra measurement by gamma-ray spectrometry may be performed by either directly analyzing the 186.2 keV photopeak of 226Ra or indirectly by analyzing the photopeaks of the radon daughters 214Bi and 214Pb. Direct measurement has the advantage of immediate counting of the sample. However, low emission probability of 186.2 keV, presence of interfering with the 185.7 keV photopeak of 235U, potential bias because of this situation and the high background rate of 186.2 keV leading to poor detection limit are the significant drawbacks of the direct measurement, especially for the low-activity concentration cases [4, 11]. On the other hand, analyzing the photopeaks of the radon daughters 214Bi and 214Pb requires the existence of secular equilibrium between 226Ra and 222Rn, which needs a waiting period of about one month before counting [13, 14]. Our experience and a number of studies in literature showed that both direct and indirect analyzing of low level measurement of 226Ra by gamma-ray spectrometry produce high relative bias between the experimental and theoretical values, and the results of the measurements are less accurate and precise compared to the results obtained by alpha-particle spectrometry [3, 5]. 228Ra can indirectly be measured by gamma-ray spectrometry via its progeny 228Ac. Since 911.1 keV photopeak of 228Ac has higher emission probability and no interference, more precise and accurate results can be expected compared to 226Ra measurement by gamma-ray spectrometry [4, 15].
The uniqueness of this study is the simultaneous measurement of 226Ra and 228Ra at the same time by using gamma-ray spectrometry and/or, depending on the 226Ra activity concentration, 226Ra measurement by alpha-particle spectrometry and 228Ra measurement by gamma-ray spectrometry by using the same source. Another good point of this study is that the same radiochemical separation procedures were applied to the samples in different matrices (two water and one shrimp samples). To have this advantageous case in source preparation and measurement steps, the unique and novel radiochemical procedures, which are the modified versions of the known procedures in literature [4, 16,17,18,19], developed in our laboratory were applied. In literature, there are radiochemical methods for 226Ra and 228Ra measurement in water samples by gamma-ray spectrometry [1, 20]. However, as mentioned in the upper paragraph, gamma-ray spectrometry may not give accurate and precise results especially for low 226Ra activities if you do not have an ultra-low-level gamma-ray spectrometry and therefore alpha-particle spectrometry is required in that case. In our method, by adjusting the appropriate concentrations of the 133Ba tracer and Pb2+ carrier at the initial steps of the procedures, the source obtained at the end included both 226Ra and 228Ra and the thickness of the source was thin enough for alpha particles to overcome self-absorption. Therefore, by using the same source, 226Ra can be measured by alpha-particle spectrometry if needed. The same radiochemical method developed for the water samples was also applied for the shrimp sample for 226Ra and 228Ra measurements with the same source, which is also a novelty. The method used in this study is new, relatively simpler and cheaper for 228Ra measurement in solid samples.
The verification of the radiochemical methods applied for the 226Ra and 228Ra measurements was performed by the satisfactory measurements of the test samples of the IAEA-TEL-2019-03 Worldwide Open Proficiency Test and IAEA-TEL-2019-04 ALMERA Proficiency Test to which we participated in 2019 [21, 22].
Experimental
PT sample description
Three PT samples were received from IAEA. The details of the test samples and the required measurements are given in the following:
Sample 01: 500 g spiked water containing anthropogenic and natural gamma emitters, 90Sr, radium isotopes of 228Ra, 226Ra and 224Ra, gross alpha and beta.
Sample 02: 500 g spiked water containing anthropogenic and natural gamma emitters, 90Sr, radium isotopes of 228Ra, 226Ra and 224Ra in low activity concentrations, gross alpha and beta.
Sample 04: 200 g shrimp samples containing natural gamma emitters and 137Cs, 210Pb, 210Po, Ra-isotopes and U, gross alpha and beta.
Reagents and radioactive standards
HNO3, H2SO4, CH3COOH provided from Sigma Aldrich, and NaOH, Pb(NO3)2, BaCl2, Na2SO4 and pH indicator (0–5) provided from Merck were used for analytical procedures. Radioactive standard solutions of 133Ba, 226Ra and 228Ra provided from Eckert-Ziegler were used to prepare calibration sources. Ultra-pure water was used in the procedures where pure water was needed.
Sample preparation procedures
Exactly the same procedures were applied to sample 01 and sample 02. A sufficient amount of water was taken gravimetrically from the test sample and put into a beaker. After checking the acidity of the sample in the beaker, the pH was adjusted to ≤ 2 by using concentrated 1 M HNO3. After adding 133Ba tracer of about 30 Bq, the sample was left to mixing on magnetic stirrer at 250 rpm for about 2 h. The radiochemical procedures described in the following parts were then applied to the sample solution.
Moisture content was determined by taking 2 g of shrimp sample and drying at 85 °C until it reached a constant weight. After that, 1 mL of 133Ba tracer of 30 Bq/mL activity was added to the sample. The sample was then ashed at 600 °C for 9 h. The ashed sample was dissolved with 25 mL of HNO3 at 65% concentration and left to mixing and totally drying on a heater magnetic stirrer at 150 °C and 230 rpm. The residual was then dissolved with 500 mL of pure water. The pH of the obtained solution was adjusted to ≤ 2 by using concentrated 1 M HNO3 and then left to mixing on the heater magnetic stirrer at 80 °C and 230 rpm for 30 min. The radiochemical procedures described in the following parts were then applied to the sample solution.
Radiochemical separation procedures
The same radiochemical separation procedures detailed in this section were applied to the water samples and to the shrimp sample. 10 mL L−1 concentrated H2SO4 was added to the samples prepared by using the above procedures and then left to mixing on the magnetic stirrer at low temperature. Pb2+ (10 mg mL−1) carrier of 0.5 ml was added to the samples in that stage. After a mixing period of 1 h, the samples were left to waiting for a night in closed beakers to provide the separation of solution and precipitate by decantation. The precipitate at this stage was Pb(Ra)SO4. The remaining mixture was then centrifuged at 3000 rpm for 5 min for the decantation of the upper aqueous part. The precipitate was washed with pure water until the pH of the upper solution was neutral and then centrifugation was again performed at the same conditions. The precipitate was then dissolved with 0.1 M EDTA/0.5 M NaOH. Ba+2 carrier of 0.3 mL (0.3 mg mL−1) was added to the solution and then mixed for a while. After adding 1 drop of indicator, 1 mL of acetic acid was added to the solution to have a pH of 4–4.5. After adding of 4 mL saturated Na2SO4 and 0.3 mL of seeding BaSO4 (10 mg mL−1), the formation of Ba(Ra)SO4 complex was observed. The samples were left to waiting for at least 30 min for the complete formation of the complex. The solution was then filtrated on a polypropylene filter (Eichrom filter of 25 mm diameter and 0.1 µm pore size) by using a vacuum pump. The separation time was recorded. After that, the filter was pasted to a steel disc by using a water-based adhesive material and was waited to be measured by the detector systems to determine the 226Ra and 228Ra activity concentrations.
Chemical recovery determination
The chemical recovery was calculated by measuring the activity of 133Ba in the disc sources prepared, which was added to the samples during the sample preparation stages, by using gamma-ray spectrometry. However, to perform such measurement the gamma-ray spectrometric system should be calibrated by using a reference standard disc of 133Ba. To have a reference standard disc, firstly a 133Ba standard solution was prepared by using a known amount (activity) of 133Ba and then 1 mL of this 133Ba standard solution was added gravimetrically to the Eichrom polypropylene filter, which is dried under the infrared lamp. After it was dried, finally, the filter was pasted to a steel disc by using a water-based adhesive material. The net peak areas of 356.01 keV energy line of 133Ba in the standard disc and in the sample prepared previously were determined by gamma-ray spectrometry. The chemical recovery (Rchem) was calculated by using Eq. (1).
where NBa133-sam and NBa133-std are the peak areas of 356.01 keV energy line in the spectrums of the sample and of the standard disc, tBa133-sam and tBa133-std are the counting times of the sample and the standard disc, mBa133-sam and mBa133-std are the amounts of 133Ba in the sample and in the standard disc, respectively.
Alpha and gamma spectrometric analysis and calculation of 228Ra and 228Ra activity concentrations
228Ra decays to 228Ac by emitting a beta particle and following gamma-rays. Since 228Ra emits very low-energy gamma rays in very low-yields (13.52 keV with 1.6% yield and 26.4 keV with 0.14% yield), the analyses based on the measurements of these gamma-rays by gamma-ray spectrometry are not reliable. Therefore, for 228Ra determination, 911.2 keV gamma-ray line of its daughter nuclide 228Ac was employed in this study. The sample was kept for about 2 days before the measurement to catch the secular equilibrium between 228Ra and 228Ac. The activity concentration of 228Ac in the water sample was determined using Eq. (2).
where NAc-228 is net peak area of 228Ac, t is time of measurement in seconds, V is volume of water sample in liters (L), ε is full energy peak efficiency, fγ is emission probability of 911.2 keV gamma-ray and Rchem is chemical recovery of the 228Ra isotope.
A characterized HPGe detector system and Genie 2000 gamma analysis software of Mirion/Canberra were used for the data acquiring and evaluation [23]. The details of the peak area calculation can be found in Şahin et al. [24]. Full energy peak efficiency, ε, was calculated by LabSOCS software of Mirion/Canberra [25] and by using an efficiency curve composed by the measurement of a standard reference source. In LabSOCS the detector and source geometries were modeled in detail and the peak efficiency values depending on energies and the true coincidence correction factor of 911.2 keV gamma-ray were determined. The measurement times were 115,000 s and 255,000 s for the water samples, and 230,000 s for the shrimp sample. The gamma-ray spectra of the Sample 02 and Sample 04 are given in Figs. 1 and 2, respectively.

The gamma-ray spectrum of the Sample 02

The gamma-ray spectrum of the Sample 04
Uncertainties reported for 228Ra measurements took into account the uncertainties due to counting statistics, full energy peak efficiency, chemical recovery and emission probability, which were the main components of the uncertainty budget.
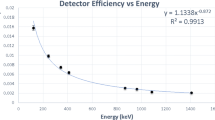
The same filter on the disc was also used to determine the 226Ra activity concentration by using alpha-particle spectrometry containing PIPS detector of 600 mm2 active area and Genie 2000 alpha analysis software. To determine the counting efficiency of the detector, the geometry factor calculation method was used by determining the solid angle [26,27,28]. The activity concentration of 226Ra in the water sample was determined using Eq. (3).
where NRa-226 is net peak area of 226Ra, t is time of measurement in seconds, V is volume of water sample in liters (L), ε is the counting efficiency and Rchem is chemical recovery of the 226Ra.
The measurement times in the alpha-particle spectrometry varied between 168,000 s – 596,000 s for the water samples and varied between 843,000 s and 864,000 s for the shrimp sample in the parallel studies for each sample. The alpha-particle spectra of the Sample 02 and Sample 04 are given in Figs. 3 and 4, respectively. The red painted areas in the spectra are the region of interest of 226Ra.

The alpha-particle spectrum of the Sample 02

The alpha-particle spectrum of the Sample 04
The main components of the combined uncertainties reported for 226Ra measurements are the counting statistics, the counting efficiency and the chemical recovery.
Data evaluation of the PT
The relative bias (RB) between the analyst’s value (Vreported) and the target value (Vtarget) is calculated by using Eq. (4).
The relative bias is compared to the Maximum Acceptable Relative Bias (MARB) which has been determined for each analyte, considering the physical background of radioanalytical methods, including the level of the activity concentration and the complexity of the task. If RB ≤ MARB, the result is the considered as “Accepted” for accuracy [21, 22].
For the evaluation of the reported result in terms of precision, the relative combined uncertainty is calculated by using Eq. (5) [21, 22]:
If the expanded relative combined uncertainty is expected to cover the relative bias, the following relation can be written as the following [21, 22]:
In the above expression, k is the coverage factor. The coverage factor is equal to 2.58 for the 99% confidential level.
The result is assigned as “Accepted” for the precision if the both P ≤ MARB and |Biasrelative|≤ k × P conditions are fulfilled. If one of the conditions is not met, the result is assigned as “Not accepted” for the precision. The final score according to the above detailed evaluation is assigned as [21, 22];
-
“Accepted” when both accuracy and precision are evaluated as “Accepted”
-
“Not accepted” when the accuracy is “Not accepted”
-
“Warning” when accuracy is “Accepted” but precision is “Not accepted”
Results and discussion
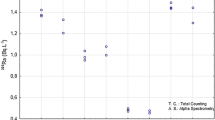
The measurement results of the three samples and the results of the performance evaluation of the reported values are presented in Table 1. The performance evaluation showed that all of the six reported values of 226Ra and 228Ra activity concentrations in one shrimp sample and two water samples passed both accuracy and precision criteria and were assigned to “accepted” status.
According to the evaluation results presented in Table 1, it can be easily said that the newly adopted radiochemical method for determining the 226Ra and 228Ra radioisotopes in water and shrimp samples worked successfully. 226Ra at high activity concentrations in samples can also be measured directly gamma-ray spectrometry. However, most of the samples such as water and shrimp have very low activity concentrations and so that measurement by gamma-ray spectrometry may not be sufficiently accurate and precise. In that case a more accurate and precise method with lower Minimum Detectable Activity (MDA), such as alpha-particle spectrometry, is needed. The MDA values for 226Ra measurement by alpha-particle spectrometry and for 228Ra by gamma-ray spectrometry were calculated as 0.3 mBq/L and 87 mBq/kg, respectively. The chemical recovery values were determined as 72% and 67% on the average for the shrimp sample and the water samples, respectively. For the alpha spectrometric measurements, the source thickness should be thin enough to discard self-attenuation. The test results showed that with this newly adopted method, sufficiently-thin sources can also be prepared in addition to the advantage of having one source for two different measurement methods.
Conclusions
The method we applied for radiochemical analyses for 226Ra and 228Ra determination in water and shrimp samples worked successfully. This unique radiochemical method developed in our laboratory including the modified procedures of the precipitation of radium with BaSO4 were performed for the measurement of 226Ra and 228Ra by using the same source with the same or different measurement techniques. The same radiochemical procedures were applied for the water and solid samples. The test samples of IAEA-TEL-2019-03 Worldwide PT, IAEA-TEL-2019-04 ALMERA PT to which we participated, were used for the verification of the method. The test results showed that the reported values of 226Ra and 228Ra in shrimp sample and two water samples with low and high activities all passed the accuracy and precision criteria and obtained “accepted” status. The relative bias values for six reported results were between 0.5 and 9.33%.
References
Jia G, Jia J (2012) Determination of radium isotopes in environmental samples by gamma spectrometry, liquid scintillation counting and alpha spectrometry: a review of analytical methodology. J Environ Radioact 106:98–119
Diab HM, Abdellah WM (2013) Validation of 226Ra and 228Ra measurements in water samples using gamma spectrometric analysis. J Water Resource Prot 5:53–57
IAEA AQ (2014) A procedure for the rapid determination of 226Ra and 228Ra in drinking water by liquid scintillation counting. IAEA analytical quality in nuclear applications series No. 39
Vasile M, Benedik L, Altzitzoglou T, Spasova Y, Watjen U, Gonzalez de Orduna R, Hult M, Beyermann M, Mihalcea I (2010) 226Ra and 228Ra determination in mineral waters—comparison of methods. Appl Radiat Isot 68:1236–1239
Al-Hamarneh IF, Almasoud FI (2018) A comparative study of different radiometric methodologies for the determination of 226Ra in water. Nucl Eng Technol 50:159–164
Hancock GJ, Martin P (1991) Determination of Ra in environmental samples by α-particle spectrometry. Appl Radiat Isot 42(1):63–69
Eikenberg J, Tricca A, Vezzu G, Bajo S, Ruethi M, Surbeck H (2001) Determination of 228Ra, 226Ra and 224Ra in natural water via adsorption on MnO2-coated discs. J Environ Radioact 54(1):109–131
Wallner G, et al. (2008) Determination of natural radionuclides in drinking water from Waldviertel, Austria. In: Proceedings of advances in liquid scintillation spectrometry. Tucson, Arizona, USA
Chalupnik S, Lebecka J (1993) Determination of 226Ra, 228Ra and 224Ra in water and aqueous solutions by liquid scintillation counting. In: Proceedings of Advances in Liquid Scintillation Spectrometry. Tucson, Arizona, USA
Repinc U, Benedik L (2001) Natural radioactivity in drinking and mineral water, In: Proceedings of advances in liquid scintillation spectrometry. Tucson, Arizona, USA
Köhler M et al (2002) Comparison of methods for the analysis of 226Ra in water samples. Appl Radiat Isot 56(1–2):387–392
Burnett WC, Tai WC (1992) Determination of radium in natural waters by liquid scintillation. Anal Chem 64:1691–1697
Michel J, Moore WS, King PT (1981) γ-Ray spectrometry for determination of radium-228 and radium-226 in natural waters Anal. Chem 53(12):1885–1889
Sidle WC, Shanklin D, Lee PY, Roose DL (2001) 226Ra and 228Ra activities associated with agricultural drainage ponds. J Environ Radioact 55:29–46
Ruberu SR, Liu YG, Perera SK (2005) Occurrence of 224Ra, 226Ra, 228Ra, gross alpha, and uranium in California groundwater. Health Phys 89(6):667–678
Sill CW (1983) Determination of radium-226 by high resolution alpha spectrometry. Report CONF-830695-Y
Durecová A, Durec F, Bursová D (2006) Determination of 226Ra, 224Ra, 223Ra and 228Ra in mineral water samples of the Slovak Republic Czech. J Phys 56(Suppl. 4):D247–D255
Thakkar AH, Fern MJ, D. McCurdy D (2003) A rapid determination of Ra-226 and Ra-224 using extraction chromatography. In: Proceeding of 9th international symposium on environmental chemical analysis, Maidstone. Special Publication –R. Soc. Chem., Cambridge. pp. 69–75
Benedik L, Spasova Y, Vasile M, Watjen U (2008) Determination of 234U, 238U and 226Ra in bottled drinking water by alpha spectrometry. In: CD Proceedings of the 7th international conference on nuclear and radiochemistry (NRC-7). Budapest, Hungary
Rihs S, Condomines M (2002) An improved method for Ra isotope (226Ra, 228Ra, 224Ra) measurements by gamma spectrometry in natural waters: application to CO2-rich thermal waters from the French Massif Central. Chem Geol 182(2–4):400–421
Mauring A, Seslak B, Tarjan S, Trinkle A (2019) IAEA-TEL-2019-03 Worldwide open proficiency test exercise, Pie-charts, S-Shapes and Reported Results with Scores
Mauring A, Seslak B, Tarjan S, Trinkle A (2019) IAEA-TEL-2019–04 ALMERA proficiency test exercise, pie-charts, S-shapes and reported results with scores
Genie 2000 (2009) Customization tools manual. Mirion technologies (Canberra Industries)
Şahin M, Dirican A, Şahin NK (2017) Radiochemical separation and determination of radium-228 in bottled mineral waters by low level gamma spectrometry and its committed effective dose. Environ Earth Sci 76:795
LabSOCS Calibration software data sheet (2017) Mirion technologies (Canberra Industries)
Pomme S (2001) Internal report IRMM. GE/R/RN/08/01
Pomme S, Paepen J (2007) A series expansion of Conway’s generalized solid-angle formulas. Nucl Instrum Meth A 579:272–274
Pomme Sl, Johansson I, Sibbens G, Denecke B (2003) An algorithm for the solid angle calculation applied in alpha-particle counting. Nucl Instrum Meth A 505:286–289
Acknowledgements
We thank to International Atomic Energy Agency for organizing Worldwide and ALMERA tests and for giving us a chance to verify our methods.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Şahin, M., Dikmen, H. Validation of a newly adopted chemical method for determination of 226Ra and 228Ra activities in water and shrimp samples. J Radioanal Nucl Chem (2023). https://doi.org/10.1007/s10967-023-09126-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10967-023-09126-6