
Abstract
Two donor-acceptor electrochromic copolymers containing fused rigid units of cyclopentadithiophene and diketopyrrolopyrrole, namely PCPDTT-DPP with benzene and thiophene ring linkers, were successfully synthesized. The electrochromic properties of the copolymers were characterized by cyclic voltammetry, UV-Vis spectroscopy and spectroelectrochemical method. The results demonstrated that the effect of DPP on the copolymer showed the p-type doping with an electrochemical band gap of 1.45 eV. The copolymer showed a higher color contrast changing from purple to transparent with %ΔT of 55% at ±1.0 V vs. Ag/AgCl compared to the parent polymer containing only cyclopentadithiophene unit. The results indicated that the new copolymer could be a potential candidate for electrochromic materials in smart films and windows.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Electrochromic polymers have been developed for smart windows, vehicle mirrors, sunglasses and displays [1,2,3,4]. The color of polymers is reversibly changed under applied voltages through an electrochemical doping process [5, 6]. A low voltage of approximately 1.0 V vs. Ag/AgCl or less with a switching time between 1.0–5.0 s is recommended in order to prolong the lifetime of the polymers. To be able to dope the polymer film at a low voltage, an energy band gap of the polymers has to be in a range of 0.5–2.0 eV [7, 8]. Another requirement to construct a high performance electrochromic device is a good color contrast of 40% or higher with an excellent switching reproducibility [9, 10].
These properties could be tuned and achieved by enhancing planarity of the polymers, copolymerizing electron donor-acceptor units and increasing the degree of conjugation via a synthetic approach [11,12,13,14,15,16]. Specifically, in the polymer backbone, the copolymerization of donor and acceptor units results in inter- or intra-molecular charge transfer bands through a push-pull interaction between donor and acceptor units resulting in unique electrical and optical properties [11, 12, 14, 16]. When the hybridization of the electronic levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of donor and acceptor matched well, it could lead to a lower band gap and favourable electrochromic properties of the polymers [17].
In order to increase the planarity of the polymer while enhancing the inter- or intra-molecular charge transfer through a push-pull interaction, a donor and an acceptor unit of fused planar conjugated unit has been reported [18,19,20,21]. A donor unit cyclopentadithiophenes (CPDT) has been shown to increase the performance of devices for solar cells [18, 19, 22,23,24,25] and electrochromics [25,26,27,28,29]. CPDT has a planar rigid structure of two fused thiophene rings which increase the degree of π-conjugation and charge transport in polymer chains relative to those of polythiophene derivatives [24,25,26, 30]. An example of an acceptor unit that has rigid fused rings and form π-π stacking is a diketopyrrolopyrrole (DPP) unit. Previous reports shows that DPP lowers a band gap of a copolymer and increases the performance of solar cells [21, 31] and electrochromics [13, 32]. Recently, several research groups reported donor-acceptor electrochromic polymers containing an acceptor DPP unit and a donor unit, which is triphenylamine [33], carbazole [34], dialkoxythiophene [35] and propylenedixoythiophene [36]. They showed that the incorporation of a DPP unit to a polymer chain improved the electrochromic properties of polymer films (i.e. fast switching time, high optical contrast and low applied voltages). However, the optical contrast of these polymer films is only about 20–30% with an application of voltage between 1.0 to 1.5 V vs. Ag/AgCl.
In order to improve the electrochromic properties of polymers, the design of random block copolymers containing CPDT blocks and DPP blocks with benzene and thiophene linker was proposed. Two fused rigid units of the donor and the acceptor should improve the planarity of the polymer chains for the improvement of charge transport and electrochromic properties (i.e. high optical contrast, fast switching time and high redox stability) in films. To the best of our knowledge, the electrochromic properties of a random copolymer of CPDT and DPP have not been reported so far. This random copolymer exhibited a high optical contrast of 55% compared to its parent polymer with only CPDT block. DPP units increased the molecular planarity of the copolymer chain shown by theoretical calculation and lowered an energy band gap down to 1.45 eV estimated by an electrochemical method.
Experimental part
Materials and measurements
All starting materials, solvents, and reagents for the synthesis of copolymers were purchased from Sigma Aldrich and Acros. The starting materials and reagents were used as received without further purification. All reactions were carried out under argon atmosphere. Lithium perchlorate and anhydrous acetonitrile were purchased from Acros and used as received. Glass substrates coated with Indium-Tin-Oxide (ITO) with a sheet resistance of 15 Ω cm−1 and 10 mm thick were purchased from Semiconductor Wafer, Inc. Each ITO glass substrate was cleaned by sonicating in ethanol for 15 min and then dried under a stream of N2 prior to film deposition.
1H NMR spectra of compounds were obtained on Bruker Biospin; DPX-300 and Bruker AVANCE 400 NMR spectrometer using CDCl3 as the solvent. Chemical shifts were recorded in parts per million (ppm), and splitting patterns were designated as s(singlet), d(doublet), t(triplet) and m(multiplet). The molecular weights of polymers were evaluated by a Jasco Gel permeation chromatographic (GPC) analyser and were measured against polystyrene standard. The UV/Vis spectra were recorded on a Perkin-Elmer Lambda 650 UV/Vis spectrophotometer. Electrochemical measurements were performed on a potentiostat (AUTOLAB PGSTAT302) in a glass cell by assembling three electrodes. A Pt disk with a diameter of 2.5 mm or a ITO glass substrate coated with polymer film was used as a working electrode. A Pt rod and a Ag/AgCl were used as a counter and a reference electrode, respectively. The spectroelectrochemical and switching measurements were carried out in a quartz UV cell (a path length of 1 cm). An ITO glass substrate coated with polymer film, a counter and a reference electrode were submerged into an electrolyte solution of 0.1 M LiClO4 in anhydrous acetonitrile. A transmission of the polymer film was monitored on a UV-vis spectrometer under an applied voltage from a potentiostat.
Polymer film preparation
The polymer film was prepared by a spin-coating method [37]. First, we prepared a polymer solution by dissolving 10 mg of PCPDTT-DPP or PCPDTT in 1.0 mL of chloroform. Then, the solution of 10 μL was dropped on an ITO glass substrate completely covering an active area of 2 cm × 2 cm. The spinning condition was at the rate of 1000 rpm for 30 s. The obtained polymer film was then ready for a measurement.
Synthetic procedures
3,6-Bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyyol-1,4-dione (3)
The reaction was adapted from the Stobbe condensation [38]. A mixture of potassium tert-butoxide (1.94 g, 17.3 mmol) and 4-bromobenzonitrile (2) (3.75 g, 20.5 mmol) was dissolved in 2-mehyl-2-buthanol (40 mL), and then the mixture was heated to 99 °C for 30 min. Dimethyl succinate (1) (1.38 mL, 8.24 mmol) was added dropwise into the reaction mixture over the period of 2.5 h at 99 °C and the reaction temperature was kept constant for 3 h. The reaction was then cooled down to 65 °C, and then methanol (20 mL) was added into the flask. Red suspension was filtered while hot and purified by recrystallized from hot methanol. The final product (3) was obtained as red powder with a 25% yield of 0.91 g. The compound was used in the next step without further purification due to its poor solubility in organic solvent. IR peaks were listed in a supporting information.
2,5-Didodecyl-3,6-bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrol-1,4-dione (4)
3,6-Bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyyol-1,4-dione (3) (566.3 mg, 1.27 mmol) and potassium tert-butoxide (313.5 mg, 2.8 mmol) were mixed in fresh distilled N-methyl pyrrolidone (NMP) (35 mL). The mixture was heated at 60 °C for 45 min. Bromododecane (2532.2 mg, 10.16 mmol) was slowly added into the reaction mixture and stirred at 60 °C for 18 h. After being cooled down to room temperature, toluene (150 mL) was added into the reaction flask. The reaction mixture was washed with water to remove the remaining NMP. The organic layer was dried over anhydrous Na2SO4, filtered and then concentrated using a rotary evaporator. The crude product was purified by column chromatography on silica as a stationary phase and using toluene as an eluent. The pure product was obtained as bright orange solid. Yield: 247.4 mg (25%). 1H NMR peaks were listed in a supporting information.
4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene (6)
4H-cyclopenta [2, 1-b; 3, 4-b′]dithiophene (5) (500 mg, 2.81 mmol) and potassium iodide (12.5 mg, 0.08 mmol) were dissolved in dimethylsulfoxide (DMSO) (12.5 mL). The reaction mixture was purged with argon for 5–10 min, and octylbromide (1085 mg, 5.62 mmol) was added into the reaction flask. Then, the reaction was cooled down to 5–10 °C in an ice bath. The potassium hydroxide (500 mg, 8.91 mmol) was added into the reaction mixture, and then stirred for 3 h at this temperature. After, the mixture was stirred for 15 h under argon at room temperature, DI water (50 mL) was added into the mixture and then extracted with diethyl ether (200 mL). The organic phase was separated and washed with DI water, saturated NaCl solution and saturated NH4Cl solution. The organic layer was dried over anhydrous Na2SO4. The organic solvent was removed under vacuum and the residue was purified by column chromatography using silica as a stationary phase, and hexane as an eluent. Fractions containing pure 4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene product were combined and evaporated. The product was obtained as colorless oil. Yield: 860 mg (76%). 1H NMR peaks were listed in a supporting information.
2,6-dibromo-4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene (7)
4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene (6) (200 mg, 0.5 mmol) was dissolved in 5 mL of anhydrous dimethylformamide (DMF) under argon in the dark. At 0 °C, N-Bromosuccinimide (NBS) (267 mg, 1.5 mmol) was added slowly. After added, the reaction mixture was allowed to cool down to the room temperature. Then, a resulting yellow solution was stirred at room temperature for 12 h. DI water (5 mL) was then added into the reaction mixture and the organic phase was extracted with diethyl ether (50 mL), washed with DI water, and 1% HCl solution and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure to obtain the desired product as yellow oil. The impurities were removed by column chromatography using silica as a stationary phase and hexane as an eluent. The compound (7) was obtained as colorless oil. Yield: 165 mg (59%). 1H NMR peaks were listed in a supporting information.
Poly[2,6-(4,4-bisoctyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene)-alt-thiophene]; PCPDTT
The reaction was adapted from Stille coupling reaction [39]. The mixture of 2,5-distannylthiophene (8) (223.4 mg, 0.54 mmol) and 2,6-dibromo-4,4-dioctylcyclopenta[2,1-b:3,4-b′]dithiophene (7) (305.6 mg, 0.54 mmol) and tetrakis(triphenylphosphine)palladium (54 mg, 0.047 mmol) was suspended in anhydrous toluene (50 mL). The reaction mixture was heated at reflux temperature for 3 days in the dark. After that, the reaction mixture was allowed to cool to room temperature, and then 2 N HCl was added into the reaction flask. The resulting mixture was extracted with chloroform (~300 mL). The organic phase was collected and washed with an EDTA solution, saturated NaHCO3 solution and DI water respectively. The organic layer was dried over anhydrous Na2SO4, filtered and removed with a rotary evaporator. The polymer was dissolved in CHCl3 and precipitated in a mixture of methanol: (2 N) HCl (10:1 v/v). The precipitates were purified by Soxhlet extraction with methanol, acetone, hexane and chloroform. The recovered chloroform fraction was concentrated and crystallized in methanol: (2 N) HCl (10:1 v/v). The precipitate was filtered off and dried under vacuum, resulting in the final product PCPDTT as a dark purple solid. 60 mg (27.6%) 1H NMR (500 MHz, C2D2Cl4):δ = 7.69 (d, 1H), 7.45 (d,1H), 6.97 (s, 2H), 5.08 (m, 4H, α-CH2), 1.96 (m, 4H, β-CH2), 1.77–1.09 (m, 40H, CH2), 0.75 (m, 6H, CH3).
13C NMR (125 MHz, C2D2Cl4): δ = 158.1, 134.4, 131.0, 130.9, 127.8, 127.7, 124.4, 122.67, 122.63, 122.5, 117.4, 117.3, 36.7, 31.3, 31.0, 29.2, 28.9, 28.5, 25.5, 23.8, 22.7, 21.8, 13.4, Mw = 19,200, Mn = 6450, PDI = 2.98.
Poly{[2,6-(4,4-bisoctyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene]-alt-thiophene}-co-[2,5-didodecyl-3,6-bis(4-phenyl)pyrrolo[3,4-c]pyrrole-1,4-dione-alt-thiophene]};
PCPDTT-DPP
The synthetic route for PCPDTT-DPP was carried out in the same manner as PCPDTT, by using 2,5-distannylthiophene (8) (223.4 mg, 0.54 mmol), 2,5-Didodecyl-3,6-bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrol-1,4-dione (4) (211.1 mg, 0.27 mmol), and 2,6-dibromo-4,4-dioctylcyclopenta[2,1-b:3,4-b′]dithiophene (7) (152.5 mg, 0.27 mmol) as starting materials. PCPDTT-DPP was obtained as a dark purple solid, (200 mg, 62%) 1H NMR (300 MHz, CDCl3):δ = 7.92 (d, 2H, thiophene-H), 7.83 (d, 2H, thiophene-H), 7.77–7.12 (m, 8H, Ph-H), 7.04 (s, 2H), 3.82 (t, 4H, α-CH2), 1.92 (t, 4H, CH2), 1.69 (m, 4H, CH2), 1.44–1.22 (m, 60H, CH2), 0.87 (t, 12H, CH3); 13C NMR (75 MHz, CDCl3):δ = 162, 158, 149, 136, 133, 131, 129, 128, 126, 125, 124, 53, 31, 30, 29.3, 29, 26, 22, 14, Mw = 77,300, Mn = 12,300, PDI = 6.30.
Results and discussion
Synthesis and characterization
The key electron accepting and donating monomers (4) and (7) were prepared as outlined in Scheme 1. For the monomer (4), first, the DPP-derivative monomer (3) was formed via a pseudo Stobbe condensation reaction between 4-bromo-benzonitrile and diethyl succinate in a single step reaction. The parent DPP monomer (3) was insoluble in common organic solvents due to the hydrogen bonding of a lactam unit. Then, by alkylating the amide nitrogens of the lactam unit with n-dodecylbromide resulted in the alkylated DPP monomer (4) which was soluble in common organic solvents. The alkylated DPP monomer (4) was then purified using column chromatography. After purification, bright orange polycrystalline solids of the monomer (4) were obtained. For the monomer (7), first, 4H-cyclopenta [2, 1-b; 3, 4-b′] dithiophene (5) was synthesized from the metallation reaction of 3- bromothiophene and n-buthylithium and followed with thiophene-3-carbaldehyde in anhydrous ether. The colorless oil of alkylated CPDT (6) was received after purification by column chromatography. 2,6-dibromo-4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene (7) was prepared from 4,4-dioctyl-4H-cyclopenta[2,1-b;3,4-b′]dithiophene (6) by bromination with N-bromosuccinimide. The crude product was purified with column chromatography to obtain the brominated CPDT monomer (7) in moderated yield (59%).

Synthesis route of monomers (4) and (7)
The preparation for PCPDTT and PCPDTT-DPP copolymers is outlined in Scheme 2. First, 2,5-bis(trimethylstannyl)thiophene monomer (8) was prepared from the reaction of thiophene by metallation with n-buthyllithium in anhydrous hexane and followed with trimethyltinchloride. The copolymer PCPDTT was prepared by the Stille coupling reaction of 2,5-bis-(trimethylstannyl)thiophene (8) and comonomer 2,5-didodecyl-3,6-bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrol-1,4-dione (DPP) (4) using a monomer mole ratio of 1:1 with Pd(PPh3)4. In order to control the repeatability for the synthesis of the copolymer PCPDTT-DPP, a monomer mole ratio of 0.5, 0.5, and 1 of (4), (8), and CPDT (7) was employed to the reaction via the Stille coupling reaction. The resulted copolymers were soluble in common organic solvents such as toluene, tetrahydrofuran, chloroform, and chlorobenzene. Molecular weights (Mn) of PCPDTT and PCPDTT-DPP were of 6450 g/mol with a PDI of 2.98 and 12,300 g/mol with a PDI of 6.30, respectively. The weight averages (Mw) of PCPDTT and PCPDTT-DPP were found to be 19,200 and 77,300 g/mol, respectively, determined by GPC performed using a UV-Vis detector set at the wavelengths of 300 nm. The Mn analysis of PCPDTT-DPP corresponded to an average degree of polymerization (DP) of approximately 12 PCPDTT repeating units (m) and 7 DPP repeating units (n) [40].

Synthesis routes of polymers PCPDTT and PCPDTT-DPP
The structures of the copolymers were confirmed via NMR spectroscopy. The 1H NMR spectrum of PCPDTT in C2D2Cl4 showed signals of thiophene unit around 7.69–7.45 ppm, and signal of derivative dithiophene unit of CPDT appeared as a singlet peak at 6.97 ppm. The proton chemical shift of n-octyl-substitute groups of CPDT was at 5.05 and in the region of 1.98–0.75 ppm.
The proton signals of the DPP phenyl unit of PCPDTT-DPP in CDCl3 showed signals of about 7.77–7.12 ppm, and signals of n-dodecyl-substituted groups of DPP appeared at 3.82 ppm and the of region about 1.69–0.87 ppm. The proton chemical shifts of the thiophene units and the derivative dithiophene units CPDT showed up around 7.92–7.83 ppm and 7.04, respectively. From the spectrum, the signals of thiophene and derivative dithiophene unit CPDT of PCPDTT-DPP shifted to the high field region compared with PCPDTT, due to electron-withdrawing ability of the DPP unit.
Optical properties of copolymers
The UV-Vis absorption spectra of the synthesized copolymers in CHCl3 solution and in solid film are shown in Fig. 1. Table 1 summarizes the corresponding optical data. In solution, PCPDTT exhibits a main absorption peak band at 570 nm while PCPDTT-DPP exhibits a main absorption peak at 580 nm. Compared to the literature, the absorption of poly(cyclopentadithiophene) in solution and as thin film was in a range of 530–567 nm [41]. In our case, the absorption is at 570 nm for PCPDTT and at 580 nm for PCPDTT-DPP as a result of an increase in the degree of conjugation from a thiophene ring and the planarity of a PCPDTT and DPP units.

Normalized UV-vis absorption spectra of polymers in CH3Cl (black line) and as thin films on quartz (red line): a PCPDTT and b PCPDTT-DPP
In the case of PCPDTT-DPP, we observed a small absorption band at 350–400 nm, which was assigned to the charge transfer of a donor PCPDTT and an acceptor DPP confirming the copolymerization of a polymer chain. The effect of the addition of the DPP acceptor block to PCPDTT block was a small red-shift of the absorption band of 10 nm due to the small number of DPP blocks adding to the polymer chain.
As thin film on a quartz substrate, the absorption spectra of PCPDTT and PCPDTT-DPP was similar due to the strong aggregation of the polymers [32]. PCPDTT showed a broad shoulder at 680 nm which was from an aggregation of different domains in polymer film. However, PCPDTT-DPP only showed one main absorption peak at 580 nm. This observation is a result of the π-π interaction of DPP units which can promote better film casting resulting in a homogeneous film [42].
We estimated an optical band gap of the polymer film from an onset of absorbance spectrum. The optical band gap of PCPDTT-DPP is (Eg = 1.70) which is slightly lower than the optical band gap of PCPDTT (Eg = 1.80 eV). From previous reports, the copolymerization of DPP acceptor unit and a donor unit such as thiophene and its derivative can reduce the Eg through push-pull charge transfer process [43]. In our system, we hypothesized that the copolymer of CPDT and DPP blocks with benzene and thiophene linkers formed a π-π stacking resulting in an increase of planarity in the copolymer chain. This effect can then lower an optical Eg and form a homogenous domain of polymer compared to PCPDTT polymer film with only CPDT units.
Electrochemical properties
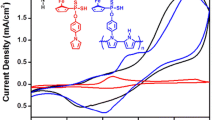
To investigate the effect of DPP units to the HOMO and LUMO energy levels of PCPDTT-DPP, we estimated the HOMO and LUMO energy levels from an onset of oxidation and reduction peaks of a cyclic voltammogram. For a comparison, the parent PCPDTT polymer was also investigated. In Fig. 2, both copolymers show a quasi-reversible oxidation process for p-type doping. In a reduction process for n-type doping, PCPDTT showed a quasi-reversible peak while PCPDTT-DPP showed an irreversible process. For both PCPDTT and PCPDTT-DPP, the color change of polymer films was only observed in p-type doping but not in n-type doping due to the instability in a n-doped state as shown by CV (Fig. 2).

CV of polymer thin films of PCPDTT and PCPDTT-DPP in 0.1 M LiClO4 in anhydrous acetonitrile at a scan rate of 50mVs−1 vs. Ag/AgCl
The HOMO, LUMO and Eg were calculated using the equations:
assuming that the known energy value of ferrocene being 4.80 eV below the vacuum level [44]. The HOMO, LUMO and Eg values are listed in Table 1. PCPDTT has an onset oxidation and an onset reduction value of 0.70 and − 0.74 V vs. Ag/AgCl, respectively, which corresponds to the HOMO energy level of −5.50 eV and the LUMO energy level of −4.06 eV leading to an electrochemical energy gap of 1.44 eV. PCPDTT-DPP has an onset oxidation and reduction potential of 0.73 and − 0.72 V vs. Ag/AgCl, respectively. These lead to the HOMO energy level of −5.53 eV, the LUMO energy level of −4.08 eV with electrochemical energy gap of 1.45 eV. For PCPDTT-DPP, the HOMO and LUMO energy levels were slightly lower in the presence of a DPP unit. The effect of the electron withdrawing was not pronounced due to weak donating phenyl and thiophene linkers in the DPP block. Therefore, the effect of the electron withdrawing DPP unit has a small effect on the energy levels observed by the electrochemical method [45, 46].
Density functional theory calculation
To further investigate effects of DPP acceptor unit on the geometry and electronic characteristics of a polymer chain, we applied the Density Functional Theory (DFT) calculations on the polymer backbones using Gaussian 09 [47]. We used functional and a basis set of B3LYP/6-31F(d,p) with PCPDTT-DPP copolymer of four units with methyl substituted side chains in the polymer skeleton. For comparison, we also applied DFT calculation to PCPDTT of three units. As shown in Fig. 3, the optimized geometry and dihedral angles of the PCPDTT trimer is linear. While PCPDTT-DPP copolymer is also quite linear from the copolymerization of PCPDTT and DPP units through benzene and thiophene linkers with a dihedral angle of 23.67°, which is relatively small [48]. It is noted that the distribution of dihedral angles throughout the polymer chain remains relatively narrow. The small dihedral angles of the PCPDTT-DPP resulted in a planarity of the polymer backbone and potentially enhance charge transport of PCPDTT-DPP.

Optimized geometry and dihedral angles of a trimer of PCPDTT and a tetramer of PCPDTT-DPP
We also obtained HOMO and LUMO energy levels for the two polymers as shown in Fig. 4. The HOMO and LUMO energy levels of PCPDTT-DPP and PCPDTT are −3.41 and − 2.82 eV and − 3.94 eV and − 3.25 eV, respectively. From the DFT calculation, the energy band gap of PCPDTT-DPP is lower than the energy band gap of PCPDTT. The HOMO of the PCPDTT-DPP unit is mostly distributed over PCPDTT unit while the LUMO of the PCPDTT-DPP unit is mostly distributed over the next PCPDTT unit and nearly absent at the DPP unit. It should be noted that the differences between the experimental and theoretical HOMO/ LUMO values can be explained by the inherent limitation of DFT calculation and the intrinsic discrepancy in the polymer backbone. DFT methods showed that DPP acceptor with benzene and thiophene linkers could lower the energy band gap of the polymer and potentially enhanced charge transport of PCPDTT-DPP.

HOMO and LUMO energy levels of a trimer of PCPDTT and a tetramer of PCPDTT-DPP
Spectroelectrochemistry
The electrochromic properties of both polymers PCPDTT and PCPDTT-DPP were investigated by monitoring transmission of the polymer films under an application of voltages. We applied a voltage from 0 to +0.9 V vs. Ag/AgCl to monitor a color change from a color state to a transparent state. With an increment of 0.1 V vs. Ag/AgCl, a voltage was applied to the polymer films. As shown in Fig. 5a, b, PCPDTT and PCPDTT-DPP films were purple in a neutral state at 570 nm and 580 nm which were assigned to be the π-π* transition electronic state. For PCPDTT film, there was no change in absorption spectrum until the voltage was increased to 0.4 V vs. Ag/AgCl. At 0.4 V, the oxidation reaction (doping process) started and the intensity of absorption peaks at 570 nm started to decrease. When the voltage was increased to 0.6 V, the color of the film turned to transparent with an increase in absorption at the wavelength above 800 nm. It indicated the formation of positive charge carriers, polarons and bipolarons at a higher energy state from an electrochemical oxidation process.

Spectroelectrochemistry of a PCPDTT and b PCPDTT-DPP on an ITO glass substrate in 0.1 M LiClO4 in anhydrous acetonitrile with an application of voltage vs. Ag/AgCl with an increment step of 0.1 V
In the case of PCPDTT-DPP films, there was no change in absorption spectrum until the voltage was increased to 0.4 V vs. Ag/AgCl and then the film turned transparent at the voltage of 0.8 V with an increase in absorption at the wavelength above 800 nm also indicating the formation of positive charge carriers polaron and bipolarons similar to the case of PCPDTT. For PCPDTT-DPP, we also observed a small absorption band at 420 nm which assigned to be a charge transfer of donor and acceptor units. After the applied voltage reaching 0.8 V vs. Ag/AgCl, the small absorption band of charge transfer between donor and acceptor units completely disappeared and the new band at the lower energy appeared at 325 nm. The appearance of this lower energy band is still under investigation.
Electrochromic switching
To investigate the switching property of the polymer films, dynamic electrochromic experiments of PCPDTT and PCPDTT-DPP films on an ITO glass substrate were performed in 0.1 M LiClO4 in anhydrous acetonitrile under an applied voltage of +1.0 and − 1.0 V vs. Ag/AgCl on an Autolab potentiostat in a double potential step chronoamperometry method with a time interval of 10 s. The transmission changes of the polymer films were monitored via a UV-Vis spectrophotometer at a wavelength of 570 nm for PCPDT and 580 nm for PCPDTT-DPP.
Figure 6 shows the results of the % transmission vs. time (s) between the neutral state and the oxidized state of both polymer films. Both polymer films showed color change to the full degree of 3–4 s in both bleach and color state. For PCPDTT-DPP, there was no transmittance change (%T) observed for the period of 400 s which indicated the favourable stabilities of the polymer film. On the other hand, the %T of PCPDTT in a color state gradually decreased overtime. It is known that polythiophene and its derivatives are susceptible to degradation due to the oxidation of the polymer chains during the generating of polaron and bipolaron species under an application of voltage with the presence of oxygen and moisture [49]. The polymer chain of PCPDTT-DPP has a higher optical contrast of 55% as compared to an optical contrast of PCPDTT of 25% under an application of voltages of +1.0 V and − 1.0 V vs. Ag/AgCl. Through the donor-acceptor electron push-pull and planarity of the polymer chain with CPDT and DPP units, the charge transport of the PCPDTT-DPP polymer chain was enhanced. As a result, under the same applied voltage, the PCPDTT-DPP polymer film has a higher optical contrast of 55% compared to PCPDTT polymer chain containing only CPDT unit.

% Transmission vs. Time (s) monitored at 570 nm for PCPDTT and at 580 nm for PCPDTTDPP. Each film on an ITO glass substrate in 0.1 M LiClO4 in anhydrous acetonitrile with an application of +1.0 V and − 1.0 V vs. Ag/AgCl with a time duration of 10 s at ambient conditions
Conclusions
A potential electrochromic copolymer of PCPDTT-DPP containing two rigid fused units of cyclopentadithiophenes (CPDT) and diketopyrrolopyrrole (DPP) were successfully synthesized. The copolymers were characterized by optical and electrochemical methods. The results were supported by DFT theoretical calculation. The results showed that DPP units increased the stability of polymer film and the optical contrast under a low applied voltage of +1.0 V/−1.0 V as compared to the polymer film without DPP acceptor unit. The addition of DPP copolymer increased the degree of planarity with an incorporation of benzene and thiophene linkers. Also, charge transfer from donor and acceptor of PCPDTT-DPP resulted in a color change under a low voltage of 0.4 V vs. Ag/AgCl and a full color change of 1.0 V. The copolymer could be a potential candidate for electrochromic polymer. This work emphasized the use of two donor-acceptor rigid units for an increase of planarity structure to enhance the charge transport in films for electrochromic applications.
References
Lang AW, Li Y, Keersmaecker MD, Shen DE, Osterholm AM, Berglund L, Reynolds JR (2018) Transparent wood smart windows: polymer electrochromic devices based on poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) electrodes. ChemSusChem 11:854–863
Li H, McRae L, Elezzabi AY (2018) Solution-processed interfacial PEDOT:PSS assembly into porous tungsten molybdenum oxide nanocomposite films for electrochromic applications. ACS Appl Mater Interfaces 10:10520–10527
Singh R, Tharion J, Murugan S, Kumar A (2017) ITO-free solution-processed flexible electrochromic devices based on PEDOT:PSS as transparent conducting electrode. ACS Appl Mater Interfaces 9:19427–19435
Yijie T, Kai Z, Zhaoyang Z, Haifeng C, Chunlin J, Yulei Z (2016) Synthesis, characterization, and electrochromic properties of donor-acceptor type polymers containing 2,1,3-benzothiadiazole and different thiophene donors. J Polym Sci Part A Polym Chem 54:2239–2246
Gunbas GE, Camurlu P, Akhmedov IM, Tanyeli C, Onal AM, Toppare L (2008) A fast switching, low band gap, p- and n-dopable donor-acceptor type polymer. J Electroanal Chem 615:75–83
Bildirir H, Osken I, Ozturk T, Thomas A (2015) Reversible doping of a dithienothiophene-based conjugated microporous polymer. Chem Eur J 21:9306–9311
Coskun Y, Cirpan A, Toppare L (2007) Construction of electrochromic devices using thiophene based conducting polymers. J Mater Sci 42:368–372
Ma K, Tang Q, Zhu C, Long J, Gong C, Fu X (2018) Novel dual-colored 1,1′,1″,1‴-tetrasubstituted (4,4′,4″,4‴-tetrapyridyl) cyclobutane with rapid electrochromic switching. Electrochim Acta 259:986–993
Ferreira LDL, Calado HDR (2018) Electrochromic and spectroelectrochemical properties of polythiophene β-substituted with alkyl and alkoxy groups. J Solid State Electrochem 22:1507–1515
Shi Y, Zhang Y, Tang K, Song Y, Cui J, Shu X, Wang Y, Liu J, Wu Y (2018) In situ growth of PEDOT/graphene oxide nanostructures with enhanced electrochromic performance. RSC Adv 8:13679–13685
Zhang Y, Kong L, Ju X, Zhao J (2018) Effects of fluoro substitution on the electrochromic performance of alternating benzotriazole and benzothiadiazole-based donor-acceptor type copolymers. Polymers 10:1–16
Jian N, Gu H, Zhang S, Liu H, Qu K, Chen S, Liu X, He Y, Niu G, Tai S, Wang J, Lu B, Xu J, Yu Y (2018) Synthesis and electrochromic performances of donor-acceptor-type polymers from chalcogenodiazolo [3,4-c]pyridine and alkyl ProDOTs. Electrochim Acta 266:263–275
Neo WT, Ye Q, Shi Z, Chua SJ, Xu J (2018) Influence of catalytic systems in Stille polymerization on the electrochromic performance of diketopyrrolopyrrole-based conjugated polymers. Mater Chem Front 2:331–337
Kong L, Wang M, Ju X, Zhao J, Zhang Y, Xie Y (2017) The availability of neutral cyan, green, blue and purple colors from simple D-A type polymers with commercially available thiophene derivatives as the donor units. Polymers 9:1–18
Gokce G, Ozkut MI (2018) An indolocarbazole based yellow-to-cyan soluble electrochromic polymer. Org Electron 52:317–322
Wu H, Qu B, Cong Z, Liu H, Tian D, Gao B, An Z, Gao C, Xiao L, Chen Z, Liu H, Gong O, Wei W (2012) A copolymer based on benzo[1,2-b:4,5-b/] dithiophene and quinozaline derivative for photovoltaic application. React Funct Polym 72:897–903
Kurowska A, Zassowski P, Kostyuchenko AS, Zheleznova TY, Andryukhova KV, Fisyuk AS, Pron A, Domagala W (2017) Effect of donor to acceptor ratio on electrochemical and spectroscopic properties of oligoalkylthiophene 1,3,4-oxadiazole derivatives. Phys Chem Chem Phys 19:30261–30276
Brebels J, Kesters J, Defour M, Pirotte G, Mele BV, Manca J, Lutsen L, Vanderzande D, Maes W (2018) A PCPDTTPD-based narrow bandgap conjugated polyelectrolyte for organic solar cells. Polymer 137:303–311
Qi F, Zhang Y, Wan M, Liu J, Huo L (2018) Enhanced photovoltaic performance of polymer solar cells through design of a fused dithienosilolodithiophene structure with an enlarged π-conjugated system. J Mater Chem C 6:4208–4216
Liang WW, Lin Y-S, Lai Y-Y, Cheng Y-J (2016) Synthesis, characterization, and photovoltaic applications of donor-acceptor alternating and random copolymers based on a ladder-type nonacyclic structure. React Funct Polym 108:113–121
Chen HY, Nikolka M, Wadsworth A, Yue W, Onwubiko A, Xiao M, White AJP, Baran D, Sirringhaus H, McCulloch I (2017) A thieno[2,3-b]pyridine-flanked diketopyrrolopyrrole polymer as an n-type polymer semiconductor for all-polymer solar cells and organic field-effect transistors. Macromolecules 51:71–79
Xu H, Fu X, Cheng X, Huang L, Zhou D, Chen L, Chen Y (2017) Highly and homogeneously conductive conjugated polyelectrolyte hole transport layers for efficient organic solar cells. J Mater Chem A 5:14689–14696
Garner LE, Viswanathan VN, Arias DH, Brook CP, Christensen ST, Ferguson AJ, Kopidakis N, Larson BW, Owczarczyk ZR, Pfeilsticker JR, Ramamurthy PC, Strauss SH, Boltalina OV, Braunecker WA (2018) Photobleaching dynamics in small molecule vs. polymer organic photovoltaic blends with 1,7-bis-trifluoromethylfullerene. J Mater Chem A 6:4623–4628
Hinkel F, Kim YM, Zagraniarsky Y, Schlutter F, Andrienko D, Mullen K, Laquai F (2018) Efficiency-limiting processes in cyclopentadithiophene-bridged donor-acceptor-type dyes for solid-state dye-sensitized solar cells. J Chem Phys 148:044703
Cho EC, Chang-Jian CW, Hsiao YS, Lee KC, Huang JH (2016) Influence of the bridging atom on the electrochromic performance of a cyclopentadithiophene polymer. Sol Energy Mater Sol Cells 150:43–50
Huang JH, Huang AT, Hsu CY, Lin JT, Chu CW (2012) Influence of molecular weight on silole-containing cyclopentadithiophene polymer and its impact on the electrochromic properties. Sol Energy Mater Sol Cells 98:300–307
Wu CG, Lu MI, Tsai PF (2009) Full-color processible electrochromic polymers based on 4,4-dioctyl-cyclopentadithiophene. Macromol Chem Phys 210:1851–1855
Sulas DB, London AE, Huang L, Xu L, Wu Z, Ng TN, Wong BM, Schlender CW, Azoulay JD, Sfeir MY (2018) Preferential charge generation at aggregate sites in narrow band gap infrared photoresponsive polymer semiconductors. Adv Opt Mater 6:1701138
Dominguez R, Schulz GL, dela Cruz P, Bauerle P, Langa F (2017) Cyclopentadithiophene-based co-oligomers for solution-processed organic solar cells. Dyes Pigments 143:112–122
London AE, Huang L, Zhang BA, Oviedo MB, Tropp J, Yao W, Wu Z, Wong BM, Ng TN, Azoulay JD (2017) Donor-acceptor polymers with tunable infrared photoresponse. Polym Chem 8:2922–2930
Kim A, Lee JH, Kim HJ, Choi S, Kim YU, Park CG, Jeong CH, Cho MJ, Choi DH (2018) New conjugated regular terpolymers based on diketopyrrolopyrrole-benzodithiophene and their application of thin film transistors and polymer solar cells. Synth Met 236:36–43
Lee JY, Han SY, Cho I, Lim B, Nah YC (2017) Electrochemical and electrochromic properties of diketopyrrolopyrrole-based conjugated polymer. Electrochem Commun 83:102–105
Li W, Michinobu T (2017) Electrochromic behavior of donor-acceptor polymers containing diketopyrrolopyrrole unit. J Photopolym Sci Technol 30:495–499
Zhang Y, Kong L, Ju X, Du H, Zhao J, Xie Y (2018) Synthesis and characterization of novle donor-acceptor type neutral green electrochromic polymers containing an indolo[3,2-b]carbazole donor and diketopyrrolopyrrole acceptor. RSC Adv 8:21252–21264
Neo WT, Cho CM, Shi Z, Chua S-J, Xu J (2016) Modulating high-energy visible light absorption to attain neutral-state black electrochromic polymers. J Mater Chem C 4:28–32
Feng F, Kong L, Du H, Zhao J, Zhang J (2018) Donor-acceptor-type copolymers based on 3,4-propylenedioxy-thiophene and 5,6-difluorobenzotriazole: synthesis and electrochromic properties. Polymers 10:427, 1–16
Krebs FC (2009) Fabrication and processing of polymer solar cells: a review of printing and coating techniques. Sol Energy Mater Sol Cells 93:394–412
Johnson WS, Daub GH (2011) Organic reactions. Wiley, New York
Bao Z, Chan W, Yu L (1993) Synthesis of conjugated polymer by the Stille coupling reaction. Chem Mater 5:2–3
Stefopoulos AA, Chochos CL, Bokias G, Kallitsis JK (2008) The role of intrachain and interchain interacitons of regioregular poly(3-octylthiophene) chains on the optical properties of a new amphiphilic conjugated random copolymer in solution. Langmuir 24:1103–11110
Kostyanovskiy VA, Troshin PA, Adam G, Sariciftci NS, Razumov VF (2012) Investigation of poly(cyclopentadithiophenes) as electron donor materials for organic solar cells. Energy Procedia 31:1–10
Biniek L, Schroeder BC, Donaghey JE, Gross NY, Ashraf RS, Soon YW, Nielsen CB, Durrant JR, Anthopoulos TD, McCulloch I (2013) New fused bis-thienobenzothienothiophene copolymers and their use in organic solar cells and transistors. Macromolecules 46:727–735
Li Y, Singh SP, Sonar P (2010) A high mobility P-type DPP-thieno[3,2-b]thiophene copolymer for organic thin-film transistors. Adv Mater 22:4862
Pommerehne J, Vestweber H, Guss W, Mahrt RF, Bassler H, Porsch M, Daub J (1995) Efficient two layer LEDs on a polymer blend basis. Adv Mater 7:551–554
Biniek L, Schroeder BC, Nielsen CB, McCulloch I (2012) Recent advances in high mobility donor-acceptor semiconducting polymers. J Mater Chem 22:14803–14813
Qu S, Tian H (2012) Diketopyrrolopyrrole (DPP)-based materials for organic photovoltaics. Chem Commun 48:3039–3051
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, Revision C.01. Gaussian Inc., Wallingford
Venkateshvaran D, Nikolda M, Sadhanala A, Lemaur V, Zelazny M, Kepa M, Hurhangee M, Kronemeijer AJ, Pecunia V, Nasrallah I, Romanov I, Broch K, McCulloch I, Emin D, Olivier Y, Cornil J, Belijonne D, Sirringhaus H (2014) Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515:384–388
Nair VS, Sun J, Qi P, Yang S, Liu Z, Zhang D, Ajayaghosh A (2016) Conjugated random donor-acceptor copolymers of [1]benzothieno[3,2-b]benzothiphene and diketopyrrolopyrrole units for high performance polymeric semiconductor applications. Macromolecules 49:6334–6342
Acknowledgements
This work was supported by the National Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Thailand. The authors would like to thank Prof. Thawatchai Tuntulani and Prof. Supapan Seraphin for fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 85 kb)
Rights and permissions
About this article

Cite this article
Jiramitmongkon, K., Chotsuwan, C., Asawapirom, U. et al. Cyclopentadithiophene and Diketo-pyrrolo-pyrrole fused rigid copolymer for high optical contrast electrochromic polymer. J Polym Res 27, 17 (2020). https://doi.org/10.1007/s10965-019-1989-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-019-1989-9