
Abstract
In this study, a new ferrocenyldithiophosphonate functionalized; [O-1-(4-phenyl)-1H-pyrrole ferrocenyldithiophosphonate (TPFc)] monomer and its conductive copolymers with pyrrole and 4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl)butane-1-amine were synthesized, characterized and their electrochromic properties were investigated. The spectroelectrochemical analyses demonstrated that the copolymer films reveal a reversible cycling with distinctive color changes in their reduced/oxidized forms. For the copolymer films P(TPFc-co-Py)/DCM, P(TPFc-co-Py)/CH3CN and P(TPFc-co-TPA)/DCM, the maximum optical contrasts (ΔT %) were measured as 28, 12 and 27 % at 800, 900 and 900 nm, respectively. Spectroelectrochemical analysis also revealed that all copolymers prepared with TPFc have lower band gaps compared to those of corresponding polymers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In previous years interest in organometallic materials has increased exceedingly in different applications. These involve energy storage systems [1], sensors [2], corrosion protection [3], electroanalysis [4], electrocatalysis [5] and electrochromic polymers and electrochromic display devices [6, 7]. In literature different kinds of conducting polymers (CP) containing metal complexes on polymer backbone were synthesized. Many CP containing metal complexes on polymer backbone were synthesized in previously studies. Conducting thin films where a metal is present in the polymer backbone are exemplified in a range of several complexes. Polythiophene-Ru(bpy) n+3 hybrids [8], ruthenium oligothienylacetylide complexes [9], oligothienylferrocene complexes [10] were studied. Between diverse organometallics, ferrocene is known for its high stability; hence for this reason it is specially preferred and valued in most of the precursor researches. Ferrocene has long been known as having excellent potential in this area and it was only shortly after its invention in 1951 that attempts were made to produce polymeric derivatives of this remarkable compound. In 1955 the first well-characterized polymers bearing ferrocene as a side-chain were obtained by the polymerization of vinyl ferrocene [11]. However, subsequent attempts to incorporate ferrocene in the main-chain were not particularly successful and it was not until the mid-1970s that well-defined ferrocene-containing polymers were finally being reported [12–14]. After those days, a concern in ferrocene functionalized CP was developed since these materials reveal low redox potential, high electron-donor ability and they generally show the redox properties of both ferrocene and the conducting polymer [15]. Lately, concern in ferrocene functionalized CP is evoked since these types of polymers show the redox properties of both groups [16]. As a result, these are useful in a range of applications and studies. CP containing ferrocene on main conjugation path of polymer backbone were also synthesized [17–19].
In this study, we disclose the synthesis and characterization of electronic properties of a novel ferrocenyldithiophosphonate-functionalized conducting polymer based on O-1-(4-phenyl)-1H-pyrrole ferrocenyldithiophosphonate (TPFc) in detail. Additionally, this study deals with electrochemical properties of novel metal-containing conducting copolymer based on TPFc with pyrrole (Py) and 4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl) butan-1-amine (TPA). Electrochromic properties of polymers were examined by spectroelectrochemistry and switching studies.
2 Experimental
2.1 Chemicals
4-(1-H-Pyrrole-1-yl)phenol (PF), acetonitrile (CH3CN), thiophene (C4H4S), toluene (C7H8), succinyl chloride (C4H6Cl2O), hydrochloric acid (HCl), sodium bicarbonate (NaHCO3), magnesiumsulphate (MgSO4), ethanol (C2H4OH), butane-1,4-diamine (C4H12N2), propionic acid (C3H6O2), tetrabutylammoniumhexafluorophosphate (TBAPF6) were purchased from Aldrich. Pyrrole is commercially available from Alfa Aesar. Dichloromethane (CH2Cl2) and AlCl3 were provided by Merck. The monomer; 4-(2,5-di(thiophene-2-yl)-1H-pyrrole-1-yl)butane-1-amine (TPA) was synthesized according to a procedure described previously [20].
2.2 Instruments
Three-electrode cell geometry was used in all electrochemical experiments. The indium tin oxide (ITO) coated glass plates of thickness of 0.7 mm with resistance of 8–12 Ω sq−1 were purchased from Delta Technologies Limited, USA, and were used as the working electrodes. Coated working electrode area arranged as 1 cm2. Pt and Ag wires were used as the counter and pseudo reference electrodes [calibrated against Fc/Fc+ (0.3 V)]. An iviumpotentiostat/galvanostat interfaced with a personal computer was used in all electrochemical measurements. Spectroelectrochemical measurements were carried out in a three-electrode quartz cell. The spectra were collected with a Diode Array UV–vis spectrophotometer (Agilent 8453) with a PC interface. The structure of the monomer was confirmed by NMR and IR spectral analysis. 1H NMR spectra of the monomer were taken by using a Bruker-instrument NMR spectrometer (DPX-400) with CDCl3 as the solvent.
2.3 Synthesis of Monomers
2.3.1 Synthesis of TPFc
TPFc was synthesized by the reaction of, [FcP(=S)(μ-S)]2 with 4-(1-H-pyrrole-1-yl)phenol in toluene (Scheme 1). Detailed synthesis route is given below:

The synthetic route for TPFc
[FcP(=S)(m-S)]2 (0.25 g, 0.446 mmol) was reacted with the 4-(1-H-pyrrole-1-yl)phenol (0.142 g, 0.892 mmol) in a 1:2 ratio in toluene (10 mL) to give O-1-(4-phenyl)-1H-pyrroleferrocenyldithiophosphonate. The reaction mixture was heated until all solids were dissolved and a yellow solution was obtained. The yellow–orange crystalline product was filtered, washed with n-hexane and dried under vacuum. Yield: 0.40 g (39.6 %), m.p.: 158 °C.
Structure of the TPFc was characterized by 1H NMR, 13C NMR and 31P NMR analysis. In the 1H NMR and 13C NMR spectra, the zero chemical shift was assigned to TMS, while in the 31P NMR spectra δ = 0 corresponds to orthophosphoric acid (H3PO4).
As a result of the 31P NMR, a single peak observed at 86,384 ppm, 13C NMR and 1H NMR analysis confirm the molecular structure of the TPFc. NMR spectra of the TPFc were given in supplementary material.
2.3.2 Synthesis of TPA
The monomer TPA;4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl) butan-1-amine was synthesized from 1,4-di(2-thienyl)-1,4-butanedione and butane-1,4-diamine in the presence of catalytical amounts of propionic acid according to the procedure reported [20]. Yellow solid was obtained at the end of the reaction and structure of TPA was characterized by NMR.
2.4 Synthesis of Copolymers
TPFc was copolymerized with Py or TPA potentiodynamically in a single compartment electrolysis cell containing 0.1 M TBAPF6/DCM or CH3CN. The copolymer was deposited on ITO coated glass slide. Pt wire and Ag wire were used as the counter and reference electrodes respectively. P(TPFc-co-Py) and P(TPFc-co-TPA) were achieved in the presence of 3.4 × 10−4 M TPFc and 0.01 M Py or TPA (Scheme 2).

Schematical representation of the electrochemical copolymerization
2.5 Investigation of Spectroelectrochemistry and Switching Time
Spectroelectrochemical analyses of the polymer were carried out to understand the band structure of the product. For spectroelectrochemical studies, the copolymer films were synthesized on ITO coated glass slide. UV–vis spectra of the film were recorded at various potentials in a monomer free solution. A square wave potential step method coupled with optical spectroscopy was used to investigate the switching times and the contrast for these copolymers. In this double potential step experiment, the potential was set at an initial potential (where the conducting polymer was in one of its extreme states) for 5 s and stepped to a second potential for another 5 s, before being switched back to the initial potential. During the experiment the percent transmittance (%∆T) and switching times at λmax of the polymer were measured using a UV–vis spectrophotometer.
3 Result and Discussion
3.1 Electrochemical Properties of Polymers
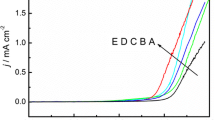
Figure 1a, b show the cyclic voltammograms of a mixture of TPFc and Py monomers and TPFc-co-Py copolymer taken in DCM/CH3CN containing 0.1 M TBAPF6. For comparison, second cycles of the CV of the TPFc, Py and TPFc-co-Py in DCM and CH3CN are given in Fig. 1a, b. Cyclic voltammograms of TPA, TPFc, TPFc-co-TPA measured with a scan rate of 0.25 V/s in DCM solution containing 0.1 M TBAPF6 are presented in Fig. 1c. As the solubility of TPA in CH3CN is low, studies were conducted with single solvent. Upon analysis of cyclic voltammograms, shapes of the voltammograms and peak potentials are seen to be different from each other. Peak potentials of the copolymer are different from those of both homopolymers indicating the formation of a copolymer. Electroactivity of the film in CH3CN was more defined with a high oxidation current compared to the one in DCM.

Cyclic voltammograms of a Py, TPFc and TPFc-co-Py in DCM, b Py, TPFc and TPFc-co-Py in CH3CN and c TPA, TPFc and TPFc-co-TPA in DCM containing 0.1 mol L−1 TBAPF6 at 250 mV/s scan rate
3.2 Electrochemical and Electrochromic Properties of Polymers
3.2.1 Cyclic Voltammetry of P(TPFc-co-Py) and P(TPFc-co-TPA)
Cyclic voltammograms of pyrrole, TPFc, TPFc-co-Py measured with a scan rate of 0.25 V/s in DCM and CH3CN solution containing 0.1 M TBAPF6 are presented in Fig. 2a–f. Oxidation and reduction peak potential values observed in the voltammetry graphics of P(Py), P(TPFc), P(TPFc-co-Py) are given in Table 1. The cyclic voltammogram of Py in DCM do not show clear redox peaks due to formation of pyrrole oligomers with different chain lengths (Fig. 2a). The potentiodynamic polymerization of Py in CH3CN is shown in Fig. 2d. During the first anodic scan, Py exhibited a single well defined peak due to the formation of the monomer radical cation. Upon repeated scanning this peak potential was decreased due to the formation of oligomers having lower oxidation potentials. Noticeable features observed from the CV studies were: the onset potentials are observed at 1.2 V for monomer oxidation in the first forward scan in both solvents but relatively higher oxidation currents were observed in CH3CN than the ones in DCM. The general characteristics of the electrochemical copolymerization and the potentials observed here for oxidation of the monomer are in good agreement with those reported previously [21]. Note that the growth pattern in DCM is totally different from that in CH3CN. Dichloromethane is a non polar solvent compared to CH3CN and thus, would dissolve the oligomers better than CH3CN, resulting in smaller rates of film growth particularly in earlier stages. This would result in an accumulation of a smaller amount of oligomers on the surface, but the smaller amounts of oligomers deposited on the surface appear to passivate the electrode as well, due to their poor electrical conductivity.

Cyclic voltammograms of a P(Py) and b P(TPFc) c P(TPFc-co-Py) in DCM containing 0.1 mol L−1 TBAPF6 d P(Py) e P(TPFc) f P(TPFc-co-Py) in CH3CN at 250 mV/s scan rate
When CV’s of the TPFc in DCM and CH3CN were analyzed, shape of the voltammograms and peak potentials are seen to be similar (Fig. 2b, e). The cyclic voltammograms of P(TPFc-co-Py) in DCM and CH3CN show a reversible redox process at about 0.45 and 0.85 V respectively referring to ferrocene group and followed by a monomer oxidation peak at 1.73 V proving the radical cation formation. Electroactivity of the film in CH3CN was more defined and has a high oxidation current when compared with CV in DCM (Fig. 2c, f).
The current of the oxidative peak progressively increases with the number of cycles indicating the formation and the growth of conducting copolymer film and suggests that there is a systematic increase in the electrode area as a result of the deposition of P(TPFc-co-Py).
The oxidation/reduction behaviors of P(TPFc-co-TPA) (0.01 M) were investigated by CV in 0.1 M TBPAF6/DCM. In order to investigate the electrochemical copolymer formation of the TPFc (0.01 M) and TPA (0.01 M), CV studies were performed under the same experimental conditions.
In the first cycle of the cyclic voltammogram, an oxidation peak emerged at 1.08 V and a reduction peak at 0.37 V. After subsequent runs electroactivity increases with increasing scan number. Shape of voltammogram and redox potentials of the TPA and TPFc mixture (Fig. 3b) were different than those of TPA (Fig. 3a) and TPFc (Fig. 2b), which in fact could be interpreted as the formation of copolymer. Oxidation and reduction peak values of P(TPA), and P(TPFc-co-TPA) which have been observed through CV are shown in Table 1.

Cyclic Voltammograms of a P(TPA) and b P(TPFc-co-TPA) in DCM containing 0.1 mol L−1 TBAPF6 at 250 mV/s scan rate
3.2.2 Scan Rate Dependence of the Peak Currents
P(TPFc-co-Py) and P(TPFc-co-TPA) films were prepared via constant potential electrolysis. Their redox switching in a monomer free electrolyte revealed a single, well-defined redox process. Figure 4 shows cyclic voltammograms of P(TPFc-co-Py) at different scan rates in DCM and CH3CN. The current responses were directly proportional to the scan rate indicating that the polymer films were electro-active and well adhered to the electrode. As seen in Fig. 4a and b the redox wave in different solvent showed the characteristic of a surface–confined redox couple, with the expected linear relationship of peak current with the potential scan rate having an anodic and cathodic least squares fit of R = 0.996, R = 0.996; R = 0.998, R = 0.999, respectively. Furthermore, the polymer film is very stable at high scan rates and the peak currents vary linearly as a function of scan rate, which confirmed a well-adhered polymer film and non-diffusional redox process (Fig. 4a, b).

Redox behaviors of a P(TPFc-co-Py) b P(TPFc-co-Py) at different scan rates in 0.1 M TBAPF6/DCM and CH3CN c P(TPFc-co-TPA) at different scan rates in 0.1 M TBAPF6/DCM
Figure 4c shows cyclic voltammograms of P (TPFc-co-TPA) at different scan rates. The scan rates for the anodic and cathodic peak currents show a linear dependence (anodic and catodic least squares fit of R = 0.999, R = 0.998, respectively) as a function of the scan rate as illustrated in Fig. 4c for P(TPFc-co-TPA). This demonstrates that the electrochemical processes are not diffusion limited.
The current responses were directly proportional to the scan rate indicating that the polymer films were electro-active and well adhered to the electrode.
3.2.3 Electrochromic Properties of the P(TPFc-co-Py) in DCM and CH3CN
The best way of examining the changes in optical properties of CP upon voltage change is via spectroelectrochemistry. It also gives information about the electronic structure of the polymer such as band gap (Eg) and the intergap states that appear upon doping.
The optoelectronic behavior of the P(TPFc-co-Py) was investigated by UV–vis spectrophotometer in a monomer free electrolyte system while incrementally increasing the applied potential between −0.2 and 1.4 V in a monomer free (0.1 M) TBAPF6/DCM medium and −0.5 and 1.0 V in a monomer free (0.1 M) TBAPF6/CH3CN (Figs. 5, 6).

Spectroelectrochemical graphic of P(TPFc-co-Py) 0.1 M TBAPF6 in DCM

Spectroelectrochemical graphic of P(TPFc-co-Py) 0.1 M TBAPF6 in CH3CN
As seen in Fig. 5 and 6, π–π* transition wavelengths (λmax) of P(TPFc-co-Py) in DCM and CH3CN were determined as 336 and 322 nm. Also the band gaps (Eg) were calculated as 1.94 and 2.12 eV, respectively. This suggests that the conjugation length of the oligomers generated during the polymerization was smaller in DCM compared to those in CH3CN. The bipolaron transition starting above 600 nm was quite strong for P(TPFc-co-Py) film prepared in DCM compared to those in CH3CN indicating that the conductivity of the doped polymer produced in DCM would also be significantly higher. This should also be due to the longer chain lengths of the film prepared in DCM.
For investigation spectroelectrochemical properties, P(TPFc-co-TPA) film was electrochemically synthesized on an ITO electrode in TBAPF6/DCM solvent/electrolyte couple. The spectroelectrochemistry studies were studied by applying potentials between −0.5 and 1.2 V in a monomer free DCM/TBAPF6 (0.1 M) medium. The wavelength (λmax) at which polymer shows π–π* transition was determined as 297 nm. The band gap (Eg) was calculated as 2.43 eV (Fig. 7).

Spectroelectrochemical graphic of P(TPFc-co-TPA) 0.1 M TBAPF6/DCM
3.2.4 Switching
The ability of a polymer to switch without any delay while exhibiting a sharp color change is very significant. Double potential step chronoamperometry was carried out to estimate the response time of the polymer film. The potential was stepped between fully oxidized and neutral states of the polymer film with a residence time of 5 s. During the experiment the percent transmittance (%∆T) at the wavelength of maximum contrast was measured using a UV–vis spectrophotometer. For P(TPFc-co-Py) maximum contrast (%∆T) and switching time were measured as 28 % and 2 s for 800 nm and 12 % and 2 s for 900 nm in DCM (Fig. 8a) and CH3CN (Fig. 8b) respectively by stepping the potential between −0.2 and 1.4 V and −0.5 and 1.0 V with a residence time of 2 s.

Potential-time, absorbance-time, current density-time for a P(TPFc-co-Py) in DCM/TBAPF6, b P(TPFc-co-Py) in CH3CN/TBAPF6 c P(TPFc-co-TPA) in DCM/TBAPF6
For the P(TPFc-co-TPA) film, the maximum optical contrasts (ΔT %) and switching time were measured as 27 % at 900 nm and 4 s by step the potential between—0.5 and 1.2 V (Fig. 8c).
Table 2 summarizes a comparison of copolymers prepared different medium.
4 Conclusion
New ferrocenyldithiophosphonate functionalized [O-1-(4-phenyl)-1H-pyrrole ferrocenyldithiophosphonate (TPFc)] monomer and its conductive copolymers with pyrrole and 4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl)butane-1-amine were synthesized, characterized and their electrochromic properties were investigated. Electrochemical behaviors of copolymers in different solvent media were examined with cycle voltammetry and spectral analysis. Our results show the role of solvents clearly in electrochemical polymerization of TPFc copolymers. The polymerization reaction and spectral properties were much more efficient in a CH3CN in comparison to that in DCM. Spectroelectrochemical analysis, switching properties, scan rate investigations and electrochromic properties of the copolymers were investigated. Spectroelectrochemical analysis revealed that the all copolymer prepared with TPFc have lower band gap compared to those of corresponding polymers.
References
J. Manuel, P. Raghavan, C. Shin, M.-Y. Heo, J.-H. Ahn, J.-P. Noh, G.-B. Cho, H.-S. Ryu, H.-J. Ahn, Mater. Res. Bull. 45, 265 (2010)
B. Lakard, G. Herlem, S. Lakard, R. Guyetant, B. Fahys, Polymer (Guildf) 46, 12233 (2005)
M.M. Gvozdenović, B.Z. Jugović, J.S. Stevanović, B. Grgur, T.L.J. Trišović, Z.S. Jugović, Synth. Met. 161, 1313 (2011)
M.R. Huang, X.W. Rao, X.G. Li, Y.B. Ding, Talanta 85, 1575 (2011)
T. Maiyalagan, J. Power Sources 179, 443 (2008)
M. Rani, R. Ramachandran, S. Kabilan, Synth. Met. 160, 678 (2010)
X. Tu, X. Fu, Q. Jiang, Z. Liu, G. Chen, Dye. Pigment 88, 39 (2011)
S.S. Zhu, R.P. Kingsborough, T.M. Swager, J. Mater. Chem. 9, 2123 (1999)
Y. Zhu, D.B. Millet, M.O. Wolf, S.J. Rettig, Organometallics 18, 1930 (1999)
Y. Zhu, M.O. Wolf, Chem. Mater. 11, 2995 (1999)
C.E. Carraher, C.U. Pittman, in Metal Containing polymeric Systems, ed. by J.E. Sheats (Plenum Press, Newyork, 1985), p. 479
A. Alkan, A. Natalello, M. Wagner, H. Frey, F.R. Wurm, Macromolecules 47, 2242 (2014)
G.K. Çilgi, M. Karakuş, M. Ak, Synth. Met. 180, 25 (2013)
J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn, M. Rehahn, Organometallics 32, 5873 (2013)
H. Gülce, A. Yetkin, E. Akgül, A. Gülce, Thin Solid Films 545, 81 (2013)
Z. Bicil, P. Camurlu, B. Yucel, B. Becer, J. Polym. Res. 20, 228 (2013)
S.J. Higgins, C.L. Jones, S.M. Francis, Synthetic Met. 98, 211 (1999)
P. Stepnicka, Ferrocenes; Ligand, Materials and Biomolecules (Chichester, John Wiley & Sons Ltd, 2008), p. 655
J. Xu, Y. Tian, R. Peng, Y. Xian, Q. Ran, L. Jin, Electrochem. Commun. 11, 1972 (2009)
İ. Yağmur, M. Ak, A. Bayrakçeken, Smart Mater. Struct. 22, 115022 (2013)
H.S. Sharma, S.-M. Park, J. Electrochem. Soc. 151, E61 (2004)
B. Ustamehmetoğlu, N. Kızılcan, Ö. Demir, Pigment Resin Technol. 41, 179 (2012)
M. Ak, B. Gacal, B. Kiskan, Y. Yagci, L. Toppare, Polymer (Guildf) 49, 2202 (2008)
Acknowledgments
This work was supported by Scientific and Technological Research Council of Turkey (TUBITAK; Project number: 111T074) and PAUBAP (2011FBE74).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soganci, T., Ak, M., Ocal, S. et al. Ferrocenyldithiophosphonate Containing Conducting Polymers and Theirs Electrochromic Application. J Inorg Organomet Polym 25, 1011–1018 (2015). https://doi.org/10.1007/s10904-015-0202-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-015-0202-9