
Abstract
Docetaxel is one of the most widely used anticancer agents. While this taxane has proven to be an effective chemotherapeutic drug, noteworthy challenges exist in relation to docetaxel administration due to the considerable interindividual variability in efficacy and toxicity associated with the use of this compound, largely attributable to differences between individuals in their ability to metabolize and eliminate docetaxel. Regarding the latter, the ATP-binding cassette transporter B1 (ABCB1, PGP, MDR1) is primarily responsible for docetaxel elimination. To further understand the role of ABCB1 in the biodistribution of docetaxel in mice, we utilized physiologically-based pharmacokinetic (PBPK) modeling that included ABCB1-mediated transport in relevant tissues. Transporter function was evaluated by studying docetaxel pharmacokinetics in wild-type FVB and Mdr1a/b constitutive knockout (KO) mice and incorporating this concentration–time data into a PBPK model comprised of eight tissue compartments (plasma, brain, heart, lung, kidney, intestine, liver and slowly perfused tissues) and, in addition to ABCB1-mediated transport, included intravenous drug administration, specific binding to intracellular tubulin, intestinal and hepatic metabolism, glomerular filtration and tubular reabsorption. For all tissues in both the FVB and KO cohorts, the PBPK model simulations closely mirrored the observed data. Furthermore, both models predicted AUC values that were with 15 % of the observed AUC values, indicating that our model-simulated drug exposures accurately reflected the observed tissue exposures. Overall, our PBPK model furthers the understanding of the role of ABCB1 in the biodistribution of docetaxel. Additionally, this exemplary model structure can be applied to investigate the pharmacokinetics of other ABCB1 transporter substrates.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Docetaxel (Taxotere®) is one of the most widely used anticancer agents. This compound, the first semisynthetic taxoid, was initially approved for use in 1996 for the treatment of metastatic breast cancer. Since then, docetaxel has been approved for a variety of indications, including common cancers such as breast, prostate and lung, as well as less common malignancies, such as gastric and head and neck cancer. While this taxane has proven to be an effective chemotherapeutic drug, noteworthy challenges exist in relation to docetaxel administration due to the considerable interindividual variability in efficacy and toxicity associated with the use of this agent [1]. The pharmacokinetic and pharmacodynamic variability of docetaxel is largely attributable to differences between individuals in their ability to metabolize and eliminate this compound. Docetaxel metabolism is primarily through the cytochrome P450 family member CYP3A4 and the ATP-binding cassette transporter B1 (ABCB1, PGP, MDR1) is responsible for elimination [2–4].
ABCB1 is a membrane-localized, energy-dependent drug efflux ATP-binding cassette (ABC) transporter with very broad substrate specificity. In mice, there are two genes that encode drug-transporting ABCB1, namely mdr1a and mdr1b [5–7]. The mouse mdr1a gene is predominantly expressed in intestine, liver, and blood capillaries of brain and testis whereas the mdr1b gene is principally found in the adrenal gland, placenta, ovaries and pregnant uterus [8]. Both mdr1a and mdr1b are expressed in the kidney [8]. In contrast, humans have only one isoform of ABCB1, which is prominent in the brush border of renal proximal tubules, in the biliary membrane of hepatocytes, in the apical membrane of mucosal cells in the intestine, in capillary endothelial cells of the brain and testis, in the adrenal gland and in placental trophoblasts [9–11]. In both mice and humans, ABCB1 functions to export xenobiotic compounds into the urine, bile and intestinal lumen and to prevent the accumulation of toxic agents in tissues such as the brain, testis and placenta. Additionally, expression of ABCB1 in tumor cells is known to confer drug resistance by pumping anticancer drugs out of the cell [12, 13].
To further understand the role of ABCB1 in the biodistribution of docetaxel in mice, we utilized physiologically-based pharmacokinetic (PBPK) modeling in mice that included ABCB1-mediated transport in relevant tissues. This type of pharmacologic modeling is a useful tool that facilitates the prediction of target tissue drug concentrations by incorporating mathematical descriptions of the uptake and disposition of chemicals based on quantitative interrelations among the critical determinants of physiological processes (i.e., absorption, distribution, metabolism and excretion) [14]. Accordingly, PBPK models are comprised of compartments corresponding to discrete tissues or groupings of tissues with appropriate volumes, blood flows, and pathways for xenobiotic clearance including pertinent biochemical and physiochemical constants [15]. Each compartment in the model is described with a mass-balance differential equation whose terms mathematically represent biological processes; the set of equations is then solved by numerical integration to simulate tissue time-course concentrations of chemicals and their metabolites [15]. The value of PBPK modeling is becoming increasingly apparent and this approach is now intensively used throughout the process of drug discovery and development [16, 17].
The PBPK model of docetaxel presented herein is comprised of eight tissue compartments (plasma, brain, heart, lung, kidney, intestine, liver and slowly perfused tissues) and, in addition to ABCB1-mediated transport, incorporates intravenous drug administration, specific binding to intracellular tubulin, intestinal and hepatic metabolism, glomerular filtration and tubular reabsorption. To evaluate the contribution of ABCB1 to the biodistribution of docetaxel, wild-type FVB and Mdr1a/b constitutive knockout (KO) mice were studied [18].
Materials and methods
Chemicals
Docetaxel (Winthrop U.S.) was acquired from the University of Colorado Hospital Pharmacy. All other reagents were of analytical grade.
Animals
Five- to six-week-old female FVB mice and four- to eight-week-old female Mdr1a/b constitutive knockout (KO) mice were purchased from Taconic. Animals were housed in polycarbonate cages and kept on a 12 h light/dark cycle. Food and water were given ad libitum. Upon arrival, mice acclimated for a minimum of seven days prior to any experimentation. All experimental procedures were approved by Colorado State University’s Animal Care and Use Committee and the Department of Defense US Army Medical Research and Material Command (USAMRMC) Animal Care and Use Review Office (ACURO).
Docetaxel pharmacokinetic studies in mice
A time course tissue and feces distribution study of docetaxel was conducted in both FVB and KO mice. Docetaxel was acquired as an initial solution of 20 mg/mL in 50/50 (v/v) ratio polysorbate 80/dehydrated alcohol, further diluted to a solution of 0.6 mg/mL in 0.9 % sodium chloride and administered via intravenous tail vein injection as a single bolus dose of 3 mg/kg. Subsequently, three mice from each cohort were sacrificed at 1, 2, 4, 8 and 12 h post docetaxel injection by cardiac stick exsanguination under isoflurane anesthesia. Plasma, brain, liver, proximal small intestine, kidney, heart, and lung tissue were immediately collected, rinsed with phosphate buffered saline, frozen in liquid nitrogen and stored at −80 °C until analysis. Feces was collected from the mice sacrificed at 12 h. For this purpose, mice from each cohort (n = 3) were housed together and the pooled feces were collected for the duration of the study. In addition, feces below the cecum were also collected upon sacrifice. All fecal samples were stored at −80 °C until analysis.
Docetaxel high-pressure liquid chromatography-tandem mass spectrometry analysis
Analysis of docetaxel in plasma and tissues was done using high-pressure liquid chromatography-tandem mass spectrometry (HPLC/MS/MS) analysis based on a method previously developed in our laboratory [19, 20] modified as follows. Briefly, docetaxel was extracted from plasma by adding 1,000 μL of ethyl acetate to 100 μL of unknown sample plasma, vortexing for 10 min and centrifuging at 18,000×g for 10 min at 4 °C. 800 μL of the organic phase was collected and evaporated to dryness using a rotary evaporator. Dried samples were reconstituted in 200 μL of 80/20 0.1 % formic acid in water/acetonitrile, vortexed for 10 min and centrifuged at 18,000×g for 10 min at 4 °C. An aliquot of 60 μL of the supernatant was injected into the LC/MS/MS system for analysis.
Tissues were homogenized at 100 mg/mL in water and 100 μL of the homogenate was extracted using the method for plasma detailed above. Fecal samples were lyophilized and drug was extracted by homogenizing the lyophilized feces at 25 mg/mL in ethyl acetate. 1,000 μL of this feces mixture was then analyzed using the method for plasma and tissues illustrated above. Standards and quality control samples were prepared in the appropriate matrix and analyzed as described above.
The HPLC system consisted of an Agilent 1200 Series binary pump SL, vacuum degasser, thermostatted column compartment SL (Agilent Technologies, Santa Clara, CA, USA) and a CTC Analytics HTC PAL System autosampler (Leap Technologies, Carrboro, NC, USA). The HPLC column was a Waters Sunfire C8 column (2.1 × 150 mm I.D., 5.0 μm bead size) (Waters Corporation, Milford, MA, USA) protected by a SecurityGuard™ C18 cartridge (4 × 2.0 mm I.D.) (Phenomenex, Torrance, CA, USA) and maintained at room temperature. The mobile phase consisted of an aqueous component (A) of 0.1 % formic acid in Milli-Q water and an organic component (B) of acetonitrile. The 4.0 min run consisted of the following linear gradient elution: 50 % A and 50 % B at 0 min, 50 % A and 50 % B at 0.5 min, 2 % A and 98 % B at 1.25 min, 2 % A and 98 % B at 3.0 min, 50 % A and 50 % B at 3.5 min and 50 % A and 50 % B at 4.0 min. The system operated at a flow-rate of 0.5 mL/min.
Mass spectrometric detection was performed on an API 3200™ triple quadrupole instrument (Applied Biosystems Inc, Foster City, CA, USA) using multiple reaction monitoring (MRM). Ions were generated in positive ionization mode using an electrospray interface. Docetaxel compound-dependent parameters were as follows: declustering potential (DP): 21 V; entrance potential (EP): 4.5 V; collision cell entrance potential (CEP): 71 V; collision energy (CE): 23 V and collision cell exit potential (CXP): 3.5 V. Source-dependent parameters were as follows: nebulizer gas (GS1): 40 psi; auxiliary (turbo) gas (GS2): 60 psi; turbo gas temperature (TEM): 400 °C; curtain gas (CUR): 30 psi; collision-activated dissociation (CAD) gas (nitrogen): 2 psi; ionspray voltage (IS): 4,500 V and interface heater (IH): 400 °C. Peak areas obtained from MRM of docetaxel (m/z 808.5 → 226) were used for quantification.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using noncompartmental modeling performed with Microsoft Excel and standard equations for noncompartmental analysis. Areas under the concentration–time curve (AUC) were calculated using the trapezoidal rule.
PBPK model development
A PBPK model for docetaxel was developed based on a model previously described by Bradshaw-Pierce et al. [19]. The modified model presented herein incorporated intravenous drug administration, specific binding to intracellular tubulin, ABCB1 transport, intestinal and hepatic metabolism, glomerular filtration and tubular reabsorption. This flow-limited model was comprised of eight tissue compartments: plasma, brain, heart, lung, kidney, intestine, liver and slowly perfused tissues.
Physiological parameters (tissue volumes and tissue blood flows) were obtained from Brown et al. [21].
The value used for the unbound fraction of docetaxel in the blood was 0.07, as docetaxel is highly bound (93 %) to plasma proteins [22]. The arterial blood drug concentration available to all tissues was considered to be the unbound docetaxel concentration in the blood.
Tissue:plasma partition coefficients were determined by parameter estimation, optimizing the fit for both the FVB and KO observed plasma and tissue concentrations.
The tubulin binding capacity for colchicine in various tissues was determined by Wierzba et al. [23]. These values were used in the present work for docetaxel tubulin binding capacities in the represented tissues (with the exception of the slowly perfused tissue tubulin binding capacity, which was determined by parameter estimation), as both colchicine and docetaxel bind to assembled tubulin with a stoichiometry of 1 mole ligand per one mole αβ subunit [23, 24].
For docetaxel tubulin binding affinity (Kd), the number used in our PBPK model was 19 nM. This value was derived from data indicating that paclitaxel (a structurally similar compound) binds reversibly to microtubules reassembled in vitro with high affinity (Kd of 10 nM) whereas the binding affinity for docetaxel, which is slightly more water soluble, is approximately 1.9-fold higher [24, 25].
The fraction of kidney blood flow filtered at the glomerulus was calculated using a glomerular filtration rate of 0.405 and 0.275 mL/min in FVB and KO mice, respectively [26]. Assuming that 9.1 % of the cardiac output goes to the kidneys [21], 26.5 and 18.7 % of the kidney blood flow is filtered at the glomerulus in FVB and KO mice, respectively.
The first-order rate constant for tubular reabsorption in the FVB mouse model was determined by parameter estimation, optimizing the fit for the FVB observed plasma and tissue concentrations. For the KO mouse model, the first-order rate constant for tubular reabsorption was also determined by parameter estimation, optimizing the fit for the KO observed plasma and tissue concentrations.
The first-order rate constant for hepatic CYP3A4 metabolism was determined by parameter estimation, optimizing the fit for both the FVB and KO observed plasma and tissue concentrations.
To describe intestinal CYP3A4 metabolism, a first-order rate constant was used for the FVB mouse model. This value was determined by parameter estimation, optimizing the fit for the FVB observed plasma and tissue concentrations.
For the KO mouse model, Michaelis–Menten kinetics were used to describe saturable intestinal CYP3A4 metabolism. The Km and Vmax values for CYP3A4 metabolism of docetaxel to an alcohol docetaxel (M2, resulting from oxidation of the tert-butyl ester side group) were obtained from van Herwaarden et al. [27]. From this work, the Km was determined to be 600 nM. The reported Vmax was 14 pmol/min/mg microsomal protein. This in vitro number was scaled for use in vivo by converting mg of microsomal protein to grams of intestine (3.16 mg intestinal microsomal protein/g intestine [28]) and correcting units for compatibility with the model, resulting in a Vmax value of 2654 nM/hr.
ABCB1 transport was described as a saturable process in the lung, kidney, heart, brain and slowly perfused tissue compartments. The Km and Vmax values were determined by parameter estimation, optimizing the fit for the FVB observed plasma and tissue concentrations.
PBPK model equations
The rate of change of the amount of drug in a generic tissue compartment with ABCB1 transport and drug metabolism is as follows:
where A T is the amount of drug in the tissue compartment, t is time, Q T is the blood flow to the tissue compartment, C A is the arterial blood drug concentration entering the tissue compartment, C VT is the venous blood drug concentration exiting the tissue compartment, A PGP is the amount of drug transported out of the tissue compartment by ABCB1 and A MET is the amount of drug metabolized in the tissue compartment.
The rate of change of the amount of drug transported out of the tissue compartment by ABCB1 is as follows:
where Vmax T is the maximum velocity of ABCB1 transport out of the tissue compartment and KM T is the Michaelis–Menten constant (Km) for ABCB1 transport out of the tissue compartment.
The rate of change of the amount of drug metabolized by a first order reaction in the tissue compartment is as follows:
where k is a first-order rate constant and V T is the volume of the tissue compartment.
The rate of change of the amount of drug metabolized by a saturable reaction in the tissue compartment is as follows:
where Vmax M is the maximum velocity of metabolism and KM M is the Michaelis–Menten constant (Km) for metabolism.
Assuming venous equilibration and specific tubulin binding, the drug concentration in the venous blood is:
where C T is the concentration of drug in the tissue compartment, P T is the tissue:plasma partition coefficient, fu is the unbound fraction of drug in the blood, TBC T is the tubulin binding capacity of the tissue compartment and TBA is the tubulin binding affinity.
Assuming the volume of the tissue is constant, the drug concentration in the tissue is:
Computer simulation
For PBPK modeling, acslX Libero version 3.0.2.1 (The AEgis Technologies Group, Inc.) was used.
Using the Parameter Estimation Wizard as detailed in the acslX Optimum User’s Guide (Version 3.0), model outputs were fitted to our experimental data by adjusting multiple model parameters simultaneously. In acslX Libero, this was accomplished by maximizing a log likelihood function using the Nelder–Mead algorithm, which makes use of a geometrical construct called a simplex. The objective of the Nelder–Mead algorithm is to generate an initial simplex in the parameter space, then iteratively adjust the locations of the vertices such that the simplex moves toward and eventually encloses the desired minimum.
In acslX Libero Parameter Estimation language, our model values that were determined by parameter estimation correspond to the “test parameters” and our observed plasma and/or tissue data functioned as the “fitted variables”. The test parameters were varied by the solver in order to achieve the best possible fit of the simulated values of these variables to the fitted variables. For the fitted variables, the solver was allowed to vary the heteroscedasticity parameter in order to achieve the best fit to the observed data.
Statistical analysis
Statistical analysis was performed using GraphPad Prism v5.01 (GraphPad Software, San Diego, California). For the comparison of concentration means, two-tailed unpaired t tests were used.
Sensitivity analysis
A normalized sensitivity analysis was performed as described in Loccisano et al. [29] to assess the influence of each PBPK model parameter on the simulated plasma AUC for the FVB mouse model. Briefly, sensitivity coefficients were calculated with the original parameters and for those resulting from a 1 % change in each parameter value. The following equation was used to calculate the normalized sensitivity coefficient (SC):
where A is the AUC resulting from the 1 % increase in the parameter value, B is the AUC resulting from the original parameter value, C is the parameter value increased by 1 % and D is the original parameter value.
Results
Docetaxel pharmacokinetics in FVB and KO mice
A time course biodistribution study of docetaxel was conducted in female FVB and KO mice. Plasma and tissue concentrations were measured 1, 2, 4, 8 and 12 h after a single intravenous bolus dose of 3 mg/kg. In addition, total unchanged docetaxel excreted in the feces was determined for the duration of the pharmacokinetic study (12 h).
Following docetaxel administration, there was a statistically significant increase in the concentration of docetaxel at 2, 4, 8 and 12 h in the lung, kidney, heart, intestine and brain of the KO versus FVB mice (Fig. 1). Additionally, a statistically significant increase was observed in the lung and brain tissue of KO versus FVB mice at 1 h post injection. Conversely, we did not note differences in docetaxel plasma concentrations between FVB and KO mice during the study. Furthermore, liver concentrations did not vary significantly between KO and FVB mice except at 12 h, when there was a 59 % increase in the docetaxel concentration in the liver of the KO versus FVB mice.

Observed docetaxel concentrations in mouse lung, kidney, heart, liver, intestine, plasma and brain and observed total docetaxel amount in feces following an intravenous dose of 3 mg/kg. Black bars represent the data from FVB mice. White bars represent the data from Mdr1a/b knockout (KO) mice. For all observed data, error bars symbolize SD
In terms of exposure, the loss of ABCB1 transporter function in the KO mice resulted in increased exposure (AUC) in all tissues with the exception of plasma (Table 1). These increases ranged from a modest 6 % in liver to a profound 444 % in brain.
Docetaxel PBPK model simulations in FVB and KO mice
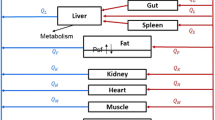
PBPK model development was based on the concentration–time data from docetaxel pharmacokinetic studies in FVB and KO mice. A schematic representation of the model is shown in Fig. 2. Values of the parameters used in this model were both mined from the literature when available and fitted to the observed plasma and tissue concentrations from the pharmacokinetic studies. As detailed in the Materials and Methods, data collected from previous work included tissue volumes and tissue blood flows, fraction of docetaxel bound to plasma proteins, tubulin binding capacities, tubulin binding affinity, glomerular filtration rate, and Km and Vmax values for intestinal metabolism in KO mice. Also described in the Materials and Methods, values determined by parameter estimation were tissue:plasma partition coefficients, the first-order rate constant for hepatic metabolism, the first-order rate constant for intestinal metabolism in FVB mice, the first-order rate constants for tubular reabsorption and Km and Vmax values for ABCB1 transport. All parameter values are listed in Table 2.

Schematic representation of a physiologically-based pharmacokinetic (PBPK) model of docetaxel incorporating intravenous drug administration, intestinal and hepatic metabolism, enterohepatic recycling (EHR), glomerular filtration, tubular reabsorption, urinary and fecal elimination and ABCB1 transport
Notably, different enzyme kinetics were used to describe intestinal metabolism in the FVB and KO mouse models. In the FVB mouse model, we represented CYP3A4 intestinal metabolism as a first-order process. Although the total amount of CYP3A in the intestine of mice is only ~2 % of that present in the liver [30] and, thus, is likely saturable at relatively low concentrations of substrate, a possible synergistic action of intestinal ABCB1 and CYP3A has been suggested that prevents enzyme saturation. Firstly, according to this mechanism, ABCB1 functions to lower the intracellular enterocyte concentration of a substrate, thereby precluding saturation of CYP3A by maintaining the substrate concentration within the linear range of the CYP3A metabolizing capacity; consequently, a larger fraction of the intracellular substrate is metabolized and overall intestinal metabolism is increased [31]. Second, the export function of ABCB1 combined with subsequent drug re-uptake results in repeated and therefore prolonged exposure of the substrate to enterocyte CYP3A, thereby increasing the probability that the substrate will be metabolized; this repeated cycling of substrate increases total metabolism, regardless of saturating or nonsaturating CYP3A kinetics [32]. Conceptually, these complex processes were incorporated into the present PBPK model with consideration for the principle of parsimony. Overall, the collective effect of the proposed synergism between intestinal ABCB1 and CYP3A is to increase intestinal metabolism. Thus, in the FVB mouse model, we eliminated ABCB1 transport out of the intestine and added compensatory first-order (nonstaurable) intestinal CYP3A metabolism kinetics. In the KO mouse model, this synergism is absent because these mice lack ABCB1; therefore, intestinal CYP3A metabolism was described by saturation (Michaelis–Menten) kinetics.
In our FVB mouse model, we also considered ABCB1 transport out of the liver to be negligible and, consequently, set the Vmax value for hepatic ABCB1 transport to zero. By setting this value to zero, we simplified complex processes for the purpose of modeling. Physiologically, while ABCB1 transporters are present in the liver, it is likely that a relatively small amount of docetaxel is actually exported because the majority of this drug is metabolized by CYP3A prior to interacting with ABCB1. An insignificant role for ABCB1 transport of docetaxel into the bile is evidence by cannulated gallbladder studies, in which equivalent amounts of unchanged docetaxel (3–4 % of the dose) were recovered in the bile of both FVB and KO mice following intravenous drug administration [33]. Similar results were observed after paclitaxel administration; in cannulated gallbladder studies, biliary excretion of unchanged paclitaxel did not differ between FVB and KO mice (5.9 and 5.2 % of the dose, respectively) [18]. Hepatic ABCB1 transport is likely minimal for the taxanes because these compounds are extensively metabolized by CYP3A4 in the liver.
Correspondingly, with our FVB mouse model void of intestinal ABCB1 transport into the lumen and hepatic ABCB1 transport into the bile, the first-order rate constant for enterohepatic recycling was zero, as the model did not allow for transport of docetaxel into the lumen. As a consequence, neither the FVB nor the KO model predicted any fecal elimination of docetaxel. This reflected our observed unchanged docetaxel recovered in the feces, which was less than 1.5 % of the administered dose in both cohorts of mice.
The concentration–time profiles of docetaxel in plasma, lung, kidney, heart, liver, intestine and brain and the resulting PBPK model simulations are shown in Fig. 3. For all tissues in both the FVB and KO cohorts, the PBPK model simulations closely mirrored the observed data.

Observed and PBPK model-simulated docetaxel concentrations in mouse lung, kidney, heart, liver, intestine, plasma and brain and model-predicted intestinal and hepatic metabolism following an intravenous dose of 3 mg/kg. Black diamonds represent the observed data from FVB mice. White diamonds represent the observed data from Mdr1a/b knockout (KO) mice. For all observed data, error bars symbolize SD. Solid lines and dashed lines indicate PBPK model predictions for FVB and KO mice, respectively
Regarding docetaxel metabolism, 77 and 80 % of the administered dose was metabolized according to the FVB and KO PBPK model simulations, respectively. The PBPK model predicted that the liver and intestine metabolized 69 and 8 %, respectively, in the FVB mice. Conversely, in the KO mice, the intestine only metabolized 2 % of the dose while the liver metabolized 78 % (Fig. 3).
The PBPK model-predicted AUCs were compared with the observed AUCs for both the FVB and KO mouse cohorts (Table 1). For this comparison, the percent difference between the observed and predicted values was calculated (Table 1). The FVB mouse model AUC predictions were all within 10 % of the observed AUCs, except for the predicted heart AUC, which was 11.3 % less than the observed AUC. For the KO mouse model, all predicted AUCs were less than 5.2 % different from the observed AUCs with the exception of plasma and brain AUCs, which were both 14.8 % greater than the observed AUCs. Overall, both models predicted AUC values that were with 15 % of the observed AUC values, indicating that our model-simulated drug exposures accurately reflected the observed exposure in lung, kidney, heart, liver, intestine, plasma and brain.
Sensitivity analysis
The normalized sensitivity coefficients for the FVB mouse PBPK model with respect to plasma AUC are shown in Fig. 4. Only parameters with sensitivity coefficients greater than 0.01 are shown. In this model, no normalized sensitivity coefficient was greater than 0.6, indicating that there are no amplified parameter errors.

Calculated sensitivity coefficients for PBPK model parameters with respect to plasma area under the concentration–time curve (AUC) for the FVB mouse model. Only parameters with sensitivity coefficients >0.01 are shown. FQ_KID fractional blood flow to kidney, FV_INT fractional volume of intestine, FV_BLD fractional volume of blood, K_LMET first-order rate constant for hepatic metabolism, KMT Michaelis–Menten constant (Km) for ABCB1 transport, VMAXT_KID maximum rate (Vmax) of ABCB1 transport from kidney, FV_KID fractional volume of kidney, FU unbound fraction of docetaxel in the blood, FQ_INT fractional blood flow to intestine
Discussion
PBPK models have been developed for numerous antineoplastic agents including methotrexate [34–36], cisplatin [37], actinomycin-D [38], 5-fluorouracil [39], capecitabine [40], 1-β-D-arabinofuranosylcytosine [41], adriamycin [42–44], topotecan [45] and docetaxel [19]. The need for these types of pharmacokinetic models for chemotherapeutics is great because of the challenges presented by this class of pharmaceutical compounds, specifically the narrow therapeutic index that is governed by drug distribution in the body. With PBPK modeling, the dynamics of drug distribution can be predicted using basic information on physiochemical properties, transport, biotransformation and excretion, thus leading to a better understanding of target tissue exposure resulting in either a therapeutic or toxic effect.
For use in PBPK model development, the role of ABCB1 in the biodistribution of docetaxel was evaluated by studying the differences in the plasma and tissue concentrations between wild-type FVB and Mdr1a/b constitutive knockout (KO) mice. Our work showed that docetaxel exposure increased by at least 100 % in the lung, intestine and brain of the ABCB1 deficient KO mice versus the wild-type mice. In contrast, plasma and liver exposure to docetaxel remained relatively unchanged between the two cohorts. These results compare closely with a similar experiment done by Kemper et al. [46], in which a statistically significant increase in exposure was found in the brain and lungs but not in the plasma or liver of KO versus FVB mice. In both studies, the largest increase in docetaxel exposure was observed in the brain (+444 and +516 % in the former and latter work, respectively).
By integrating the FVB and KO mouse tissue distribution data into a PBPK model, we were able to effectively predict docetaxel concentrations in plasma, brain, heart, lung, kidney, intestine, liver and slowly perfused tissues after an intravenous dose of 3 mg/kg. To include ABCB1 transport and metabolism into our PBPK models, we considered the lumen-to-enterocyte recycling process (the entry and exit of a compound across the intestinal epithelium multiple times which leads to in an increase in drug residency time within the enterocyte) [47–49]. The results from the development of a PBPK model which incorporated CYP3A metabolism and ABCB1 transport for the prediction of intestinal drug absorption support the notion of a drug ‘cycling’ effect that ABCB1 efflux imposes on the intestine which causes enhanced drug metabolism [48]. This PBPK model used seven luminal compartments to represent the small intestine wherein each luminal compartment was associated with a unique enterocyte compartment, with no transit of drug between adjacent enterocyte compartments. Additionally, each luminal compartment was assigned a unique ABCB1 abundance factor, CYP3A4 abundance factor, transit rate constant, absorption rate constant and basolateral to apical transfer rate constant. As docetaxel-specific rate constants necessary for the implementation of this segmented intestinal model are not yet available, we simplified the lumen-to-enterocyte recycling phenomenon in our model by eliminating ABCB1 transport out of the intestine in the wild-type mice and, as compensation, representing intestinal metabolism as a first-order, nonsaturable process. In our KO mouse model, lumen-to-enterocyte recycling is nonexistent, as these mice lack ABCB1. Thus, KO mice do not demonstrate a compensatory increase in intestinal metabolism and, consequently, CYP3A4 metabolism in the intestine was described by saturation kinetics in this cohort.
By permitting first-order intestinal metabolism in the FVB mouse model, the predicted amount of docetaxel metabolized in the intestine was 8 % whereas the liver was responsible for metabolizing 69 % of the administered dose. Physiologically, these numbers are relevant as evidenced by a study in which CYP3A4-transgenic mice were generated that either expressed CYP3A4 in the intestine or in the liver [27]. Mice with CYP3A4 expression in only the intestine were able to clear 21 % of the docetaxel cleared from the plasma by wild-type mice. Additionally, in these transgenic mice with no CYP3A4 expression in the liver, 13.5 % of docetaxel metabolites M1-4 were recovered in the small intestine relative to wild-type mice. Thus, this data demonstrates that the intestine alone is capable of metabolizing a significant amount of docetaxel and our FVB mouse model-simulated value of 8 % is physiologically plausible.
Our KO mouse model does not incorporate ABCB1 transport and the consequent lumen-to-enterocyte recycling process; therefore, with saturable (Michaelis–Menten) intestinal metabolism kinetics, the intestine was predicted to only metabolize 2.6 % of the docetaxel metabolized by the liver in these mice. This value is in accordance with the total amount of CYP3A present in the intestine of mice, which is only ~2 % of that present in the liver [30].
Overall, our pharmacokinetic study and PBPK model highlight the importance of ABCB1 transport in the biodistribution of docetaxel. As is well known, many therapeutic agents are ABCB1 substrates and, thus, likely are subject to similar pharmacokinetic changes when ABCB1 function is altered. To our knowledge, the only other whole-body mouse PBPK model that incorporates ABCB1 transport (but only in non-eliminating tissues, namely brain and heart) is a model of domperidone, an antiemetic drug associated with cardiac toxicity [50]. Both this and our work clearly illustrate the utility of PBPK modeling for further understanding the physiological mechanics of drug distribution in tissues expressing ABCB1.
An in-depth comprehension of the effects of ABCB1 transport on drug pharmacokinetics and pharmacodynamics is advantageous to human medicine because large interindividual differences in ABCB1 expression have been reported. While no null alleles have been found for ABCB1 in humans thus far, single nucleotide polymorphisms (SNPs) that affect the structure and function of the transporter have been discovered [51]. One of the most frequently found set of variants is the 1236C > T (G412G), 2677G > T (A893S) and 3435C > T (I1145I) haplotype that is found in roughly 25–40 % of Caucasians and Asians [52]. In a study comprising the pharmacogenetic screening of CYP3A and ABCB1 in relation to population pharmacokinetics of docetaxel, the homozygous 1236C > T polymorphism in the ABCB1 gene was significantly correlated with a 25 % decrease in docetaxel clearance [53]. This is in contrast to work that found polymorphisms in the CYP3A genes but not in ABCB1 had a profound effect on docetaxel exposure [54]. Thus, the former study suggests that dose-adaptation based on characterization of the 1236C > T status of ABCB1 may result in reduced interindividual variation of docetaxel pharmacokinetics while the latter study argues against screening for ABCB1 polymorphisms. However, a critical limitation of both studies is that docetaxel analysis was performed only in human plasma, which, of course, is common as it is not feasible to collect actual tissue concentration data from humans.
As shown by our work, although plasma docetaxel concentrations are virtually the same in FVB and KO mice, there are significant differences in tissue exposure to this taxane that are directly related to ABCB1 transport. And, it is in these tissues that docetaxel-associated toxicities occur. Thus, it is of the utmost importance to understand not only the plasma but also the tissue distribution of docetaxel (as well as other drugs) to truly assess the necessity of dose modifications based on protein functionality. For this purpose, PBPK modeling is an ideal tool. Our data and model suggest that adjusting the dose of docetaxel in relation to ABCB1 function is imperative to minimize detrimental tissue exposure and toxicity related to this compound. To determine the pertinence of this type of dose modification to humans, the present mouse PBPK model can be scaled to humans by taking into account interspecies differences in physiology and physiochemistry. In this way, we can estimate the affect of ABCB1 transport on both the plasma and tissue distribution of docetaxel in humans and subsequently use in silico experimentation prior to clinical trials for optimization of the administration of docetaxel to maximize efficacy and minimize toxicity.
References
Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB (1996) A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm 24(2):153–172
Bruno R, Sanderink GJ (1993) Pharmacokinetics and metabolism of Taxotere (docetaxel). Cancer Surv 17:305–313
Marre F, Sanderink GJ, de Sousa G, Gaillard C, Martinet M, Rahmani R (1996) Hepatic biotransformation of docetaxel (Taxotere) in vitro: involvement of the CYP3A subfamily in humans. Cancer Res 56(6):1296–1302
Shirakawa K, Takara K, Tanigawara Y, Aoyama N, Kasuga M, Komada F, Sakaeda T, Okumura K (1999) Interaction of docetaxel (“Taxotere”) with human P-glycoprotein. Jpn J Cancer Res 90(12):1380–1386
Gros P, Croop J, Housman D (1986) Mammalian multidrug resistance gene: complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell 47(3):371–380
Hsu SI, Lothstein L, Horwitz SB (1989) Differential overexpression of three mdr gene family members in multidrug-resistant J774.2 mouse cells. Evidence that distinct P-glycoprotein precursors are encoded by unique mdr genes. J Biol Chem 264(20):12053–12062
Devault A, Gros P (1990) Two members of the mouse mdr gene family confer multidrug resistance with overlapping but distinct drug specificities. Mol Cell Biol 10(4):1652–1663
Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE (1989) The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol 9(3):1346–1350
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC (1987) Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 84(21):7735–7738
Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR (1989) Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci USA 86(2):695–698
Sugawara I, Kataoka I, Morishita Y, Hamada H, Tsuruo T, Itoyama S, Mori S (1988) Tissue distribution of P-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody, MRK 16. Cancer Res 48(7):1926–1929
Dano K (1973) Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochim Biophys Acta 323(3):466–483
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5(3):219–234
Krishnan K, Loizou GD, Spendiff M, Lipscomb JC, Andersen ME (2010) PBPK modeling: a primer. In: Krishnan K, Andersen ME (eds) Quantitative modeling in toxicology. Wiley, Chichester, p xvii, 485
Andersen ME, Yang RSH, Clewell HJ 3rd, Reddy MB (2005) Introduction: a historical perspective of the development and applications of PBPK models. In: Reddy MB, Yang RSH, Clewell HJ 3rd, Andersen ME (eds) Physiologically based pharmacokinetic modeling: science and applications. Wiley-Interscience, Hoboken, p xix, 420
Norris DA, Leesman GD, Sinko PJ, Grass GM (2000) Development of predictive pharmacokinetic simulation models for drug discovery. J Control Release 65(1–2):55–62
Theil FP, Guentert TW, Haddad S, Poulin P (2003) Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol Lett 138(1–2):29–49
Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JM, Fibbe WE, Borst P (1997) Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA 94(8):4028–4033
Bradshaw-Pierce EL, Eckhardt SG, Gustafson DL (2007) A physiologically based pharmacokinetic model of docetaxel disposition: from mouse to man. Clin Cancer Res 13(9):2768–2776
Gustafson DL, Long ME, Zirrolli JA, Duncan MW, Holden SN, Pierson AS, Eckhardt SG (2003) Analysis of docetaxel pharmacokinetics in humans with the inclusion of later sampling time-points afforded by the use of a sensitive tandem LCMS assay. Cancer Chemother Pharmacol 52(2):159–166
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13(4):407–484
Urien S, Barre J, Morin C, Paccaly A, Montay G, Tillement JP (1996) Docetaxel serum protein binding with high affinity to alpha 1-acid glycoprotein. Investig New Drugs 14(2):147–151
Wierzba K, Sugiyama Y, Okudaira K, Iga T, Hanano M (1987) Tubulin as a major determinant of tissue distribution of vincristine. J Pharm Sci 76(12):872–875
Diaz JF, Andreu JM (1993) Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry 32(11):2747–2755
Chabner B, Longo DL (2010) Cancer chemotherapy and biotherapy : principles and practice, 5th edn. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia
Huls M, Kramers C, Levtchenko EN, Wilmer MJ, Dijkman HB, Kluijtmans LA, van der Hoorn JW, Russel FG, Masereeuw R (2007) P-glycoprotein-deficient mice have proximal tubule dysfunction but are protected against ischemic renal injury. Kidney Int 72(10):1233–1241
van Herwaarden AE, Wagenaar E, van der Kruijssen CM, van Waterschoot RA, Smit JW, Song JY, van der Valk MA, van Tellingen O, van der Hoorn JW, Rosing H, Beijnen JH, Schinkel AH (2007) Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Investig 117(11):3583–3592
Zhang QY, Dunbar D, Kaminsky LS (2003) Characterization of mouse small intestinal cytochrome P450 expression. Drug Metab Dispos 31(11):1346–1351
Loccisano AE, Campbell JL Jr, Butenhoff JL, Andersen ME, Clewell HJ 3rd (2012) Comparison and evaluation of pharmacokinetics of PFOA and PFOS in the adult rat using a physiologically based pharmacokinetic model. Reprod Toxicol 33(4):452–467
Hudachek SF, Gustafson DL (2013) Physiologically based pharmacokinetic model of lapatinib developed in mice and scaled to humans. J Pharmacokinet Pharmacodyn 40(2):157–176
van Waterschoot RA, Schinkel AH (2011) A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: recent insights from knockout and transgenic mice. Pharmacol Rev 63(2):390–410
Benet LZ (2009) The drug transporter-metabolism alliance: uncovering and defining the interplay. Mol Pharm 6(6):1631–1643
Bardelmeijer HA, Ouwehand M, Buckle T, Huisman MT, Schellens JH, Beijnen JH, van Tellingen O (2002) Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res 62(21):6158–6164
Bischoff KB, Dedrick RL, Zaharko DS (1970) Preliminary model for methotrexate pharmacokinetics. J Pharm Sci 59(2):149–154
Bischoff KB, Dedrick RL, Zaharko DS, Longstreth JA (1971) Methotrexate pharmacokinetics. J Pharm Sci 60(8):1128–1133
Zaharko DS, Dedrick RL, Bischoff KB, Longstreth JA, Oliverio VT (1971) Methotrexate tissue distribution: prediction by a mathematical model. J Natl Cancer Inst 46(4):775–784
Evans WE, Crom WR, Tsiatis A, Green AA, Hayes FA, Pratt CB (1982) Pharmacokinetic modeling of cisplatin disposition in children and adolescents with cancer. Cancer Chemother Pharmacol 10(1):22–26
Lutz RJ, Galbraith WM, Dedrick RL, Shrager R, Mellett LB (1977) A model for the kinetics of distribution of actinomycin-D in the beagle dog. J Pharmacol Exp Ther 200(3):469–478
Collins JM, Dedrick RL, King FG, Speyer JL, Myers CE (1980) Nonlinear pharmacokinetic models for 5-fluorouracil in man: intravenous and intraperitoneal routes. Clin Pharmacol Ther 28(2):235–246
Tsukamoto Y, Kato Y, Ura M, Horii I, Ishitsuka H, Kusuhara H, Sugiyama Y (2001) A physiologically based pharmacokinetic analysis of capecitabine, a triple prodrug of 5-FU, in humans: the mechanism for tumor-selective accumulation of 5-FU. Pharm Res 18(8):1190–1202
Dedrick RL, Forrester DD, Ho DH (1972) In vitro-in vivo correlation of drug metabolism–deamination of 1-D-arabinofuranosylcytosine. Biochem Pharmacol 21(1):1–16
Harris PA, Gross JF (1975) Preliminary pharmacokinetic model for adriamycin (NSC-123127). Cancer Chemother Rep 59(4):819–825
Chan KK, Cohen JL, Gross JF, Himmelstein KJ, Bateman JR, Tsu-Lee Y, Marlis AS (1978) Prediction of adriamycin disposition in cancer patients using a physiologic, pharmacokinetic model. Cancer Treat Rep 62(8):1161–1171
Gustafson DL, Rastatter JC, Colombo T, Long ME (2002) Doxorubicin pharmacokinetics: macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci 91(6):1488–1501
Sung C, Blaney SM, Cole DE, Balis FM, Dedrick RL (1994) A pharmacokinetic model of topotecan clearance from plasma and cerebrospinal fluid. Cancer Res 54(19):5118–5122
Kemper EM, Verheij M, Boogerd W, Beijnen JH, van Tellingen O (2004) Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer 40(8):1269–1274
Padowski JM, Pollack GM (2010) Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Methods Mol Biol 596:359–384
Badhan R, Penny J, Galetin A, Houston JB (2009) Methodology for development of a physiological model incorporating CYP3A and P-glycoprotein for the prediction of intestinal drug absorption. J Pharm Sci 98(6):2180–2197
Kivisto KT, Niemi M, Fromm MF (2004) Functional interaction of intestinal CYP3A4 and P-glycoprotein. Fundam Clin Pharmacol 18(6):621–626
Fenneteau F, Turgeon J, Couture L, Michaud V, Li J, Nekka F (2009) Assessing drug distribution in tissues expressing P-glycoprotein through physiologically based pharmacokinetic modeling: model structure and parameters determination. Theor Biol Med Model 6:2
Robey RW, Massey PR, Amiri-Kordestani L (2010) Bates SE ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem 10(8):625–633
Fung KL, Gottesman MM (2009) A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta 1794(5):860–871
Bosch TM, Huitema AD, Doodeman VD, Jansen R, Witteveen E, Smit WM, Jansen RL, van Herpen CM, Soesan M, Beijnen JH, Schellens JH (2006) Pharmacogenetic screening of CYP3A and ABCB1 in relation to population pharmacokinetics of docetaxel. Clin Cancer Res 12(19):5786–5793
Baker SD, Verweij J, Cusatis GA, van Schaik RH, Marsh S, Orwick SJ, Franke RM, Hu S, Schuetz EG, Lamba V, Messersmith WA, Wolff AC, Carducci MA, Sparreboom A (2009) Pharmacogenetic pathway analysis of docetaxel elimination. Clin Pharmacol Ther 85(2):155–163
Acknowledgments
We are grateful to AJ Beaupre for performing all of the intravenous tail vein injections for the mouse studies. Additionally, we are grateful to Robin McDougall (The AEgis Technologies Group, Oshawa, ON, USA) for all of his help and guidance with the PBPK modeling. This work was supported by grant number W81XWH-09-1-0457 from the Department of Defense (DOD) Breast Cancer Research Program (BCRP) of the Office of the Congressionally Directed Medical Research Programs (CDMRP).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hudachek, S.F., Gustafson, D.L. Incorporation of ABCB1-mediated transport into a physiologically-based pharmacokinetic model of docetaxel in mice. J Pharmacokinet Pharmacodyn 40, 437–449 (2013). https://doi.org/10.1007/s10928-013-9317-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-013-9317-1