
Abstract
In this study, PBT/recycled-PET blends were developed using a twin-screw extruder. A commercial chain extender (Joncryl ADR 4468) was also used to melt mix with PBT/recycled-PET blend systems. Firstly, Joncryl at different loadings was extruded with recycled-PET to explore the influence of branching on melt behavior and crystallization of PET. The effect of blending recycled-PET with PBT on the final properties was then explored at different blending ratios (25w/75w, 50w/50w and 75w/25w). Similar blends were subsequently prepared while incorporating chain extender. Melt behavior, phase miscibility, crystallization behavior, solid viscoelastic properties, tensile and impact properties of the blends were eventually analyzed using differential scanning calorimeter (DSC), melt flow indexer (MFI), dynamic mechanical analyzer (DMA), and tensile and Izod notched impact testing, respectively. The results showed that the addition of chain extender increased the melt viscosity of PET and at the low contents enhanced the PET’s crystallization rate. On the other hand, the blends of PBT/recycled-PET are fully miscible in the amorphous region whereas the crystalline phases are immiscible subsequent to a fast cooling. The PBT and PET molecules could also co-crystallize and be fully miscible in crystalline phases upon slow cooling of the melt. Blending recycled-PET with PBT didn’t suppress the tensile properties of PBT, however it could enhance the PBT’s ductility and reduce its impact strength. The chain extender didn’t influence the mechanical properties much.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(ethylene terephthalate) PET is the most commonly used commercial polyester due to its its low material cost and superior properties compared to other engineering thermoplastic polyesters. PET possesses excellent thermal and mechanical properties, high chemical resistance, high transparency, and good barrier properties against oxygen and water vapor [1,2,3]. Due to these features, PET is widely used in applications such as textile fibers, soft-drink bottles, packaging films, automotive structural components, and electrical parts [4, 5]. Among these applications, fiber-grade and bottle-grade PETs dominate the global PET market as commodity products [6]. As a consequence, there is a large amount of landfill of these daily used products and hence the wastes of PET dramatically threats the beauty of the nature and influences the global warming. The PET wastes are largely involved among almost 35% of the total plastics that are disposed in the landfills. In 2012 in Europe, 26% of total post-consumed plastics were recycled, 36% was incinerated for energy recovery however, 38% of these plastics still had to be buried in the landfill [7]. Therefore, recycling PET products is of a great importance for rescuing the environment and the earth for the future generations. It is reported by Arena et al. [8] that the production of 1 kg of recycled-PET (r-PET) flake instead of virgin PET saves approximately 30 MJ energy, which is equivalent to the energy content of about 1 l gasoline.
The use of chain extender (CE) has been shown to be a solution to improve the properties of recycled polymers, although the use of CE could sometimes be costly. In this context, the CE compensates the low molecular weight of r-PET by extending or branching the r-PET molecules and hence improves its melt strength. Therefore, rheological and melt properties of the r-PET and hence its poor processability, formability and foamability could be improved [9,10,11]. There are a wide range of commercially available CEs that could be used for PET. Depending on the nature, reactivity and number of functional groups on the CE molecules, different properties could be obtained in the final product [12]. Bi-functional CEs such as di-epoxides, di-isocyanates, di-anhydrides or bis (oxaline)s promote linear chain extension. On the other hand, tri- or tetra-functional CEs such as tetra-epoxides, tetra-functional tetra-glycidyl diamino diphenyl methane could generate branched structures in PET [10]. Several studies have been conducted to investigate the effect of addition of CE on the final properties of PET. Few studies revealed that the addition of pyromellitic dianhydride (PMDA) as the CE results in a significant decrease in hydroxyl content and an increase in intrinsic viscosity of r-PET [13]. The increase in PMDA content also increases the complex viscosity at low frequencies and improves the shear thinning behavior of r-PET [14, 15]. This is due to the increased molecular weight and increased molecular entanglement. Japon et al. [16] also studied the use of multi-functional epoxy-based CE with different degrees of functionality to increase the melt strength of PET. This CE with multi-functional epoxides induced long chain branching and caused improvements in both shear and elongational viscosities. Awaja et al. [13] showed that the crystallinity of chain extended r-PET decreased with increasing degree of branching. Other studies reported that the crystallinity depression occurs during chain extension due to the increase in molecular entanglement caused by high degree of branching and cross linking [4, 9, 17, 18]. Raffa et al. [19] illustrated that both di-functional and multi-functional CEs depress the crystallinity and crystallization rate of r-PET. On the other hand, Rosu et al. [20] claimed that the addition of branched PET to linear PET promoted the linear PET’s crystallinity. Li et al. [21] disclosed that at low degrees of branching the PET’s crystallization rate could increase as the branched sites could behave as nucleation points while the increase in branching degree suppresses the crystallization due to the reduced molecular regularities. This phenomena was also confirmed by Nofar et al. [22, 23] but on PLA samples that have also similar slow crystallization rate.
In recent years, another method to overcome the reduced properties of recycled polymers and thereby to improve the features of the final products is blending with other polymers that have superior properties with respect to those weak features of the recycled polymer. Blending recycled polymers with cheaper polymers but with appropriate features could also reduce the final cost of the products. According to their morphology, polymer blends can be classified into either miscible or immiscible. While miscible blends are single-phase down to the molecular level, immiscible polymer blends are phase separated in the microstructure. In immiscible polymer blends further compatibilization might be required to form a more stable blend with better phase interfacial interactions between the polymeric phases [24,25,26].
Blends of poly(butylene terephthalate) (PBT) and PET are commercialized thermoplastic polyester products which are mostly processed in injection molding to produce products with required heat resistance and glossy surface appearance. Similar to PET, PBT is a commercial aromatic thermoplastic polyester which is widely used in engineering applications. Chemical structures of PBT and PET are quite similar and thereby PET and PBT form stable blends without requiring compatibilizers due to their miscibility at the amorphous phase as also a single glass-transition temperature (Tg) appears intermediate between those of individual components [27]. On the other hand, PBT has a very fast crystallization rate due to the existence of flexible butylene groups within its molecular structure whereas PET possesses very slow crystallization kinetics which also causes the cold crystallization phenomena during heating cycles or annealing. As a result of this difference in crystallization rates, separate crystalline phases could form with no evidence of co-crystallization. Therefore, these blends could be immiscible in the crystalline state and double melting peaks with slightly reduced temperatures of the peaks could be observed [27]. Similar results on miscibility of amorphous and immiscibility of crystalline phases were confirmed by Avramova [28]. Since slow crystallization and poor processability of PET are somewhat PET’s main drawbacks, blending with PBT with faster crystallization rate, better processability, and with good compatibility with PET could be a breakthrough to enhance PET’s and specially r-PET’s shortcomings [29,30,31,32]. Furthermore, PBT/PET blends have been developed principally to improve processability, surface appearance, heat deflection temperature (HDT), impact strength and dimensional stability of final products [1]. These blends also benefit from the low cost of PET and the fast crystallization rate of PBT. The blends of PBT/PET could also be utilized in home appliances, electrical and automotive applications where high stiffness and strength, high heat and chemical resistance and glossy surface appearance are required [1, 33]. It has been reported that the addition of PBT could also enhance the impact strength of PET [31].
Although from the DSC thermograms, several studies have illustrated that the blends of PBT and PET are miscible, Aravinthan and Kale observed in their scanning electron micrographs that the PBT/PET blends at close blending ratios formed co-continuous and fibrillar immiscible morphologies [34]. Szostak [31] also revealed that the addition of PBT into PET increased the crystallization rate and the degree of crystallinity of the blend while decreasing the crystallization temperature. Tao et al. [35] claimed that the presence of PBT caused better crystal nucleation in PET while broadening its processing temperature.
Series of patents exist in the subject of PBT/r-PET blends [36,37,38]. They showed that after series of modifications the performance of PBT/r-PET material prepared by different formulation could be equaled to that of PBT/virgin PET. Baxi et al. [39] investigated the thermal, mechanical and microstructural properties of PBT/PET blends mixed with r-PET. They prepared their samples by addition of 60 or 90 wt% of r-PET into virgin PET/PBT blend either with composition of 40:60 or 20:80. They claimed the mechanical property and crystallinity enhancements in the blend system of 60 wt% r-PET into 40:60 virgin PET/PBT blend due to crosslinking of the polymeric chains between r-PET and blends structure. Rahmat et al. [40] studied the effect of CE content on mechanical properties and viscosity of PBT/glass-fiber reinforced r-PET composites. They used styrene-acrylic multifunctional oligomer (Joncryl ADR-4368) as the CE. They observed a significant viscosity rise as well as melt volume rate (MVR) drop after addition of 0.5 wt% and 0.65 wt% of CE into 50:50 PBT/glass-fiber reinforced r-PET. The limited dosage of CE is determined as 0.65 wt% according to their study due to the gel formation caused by high level of crosslinking.
In this study, PBT blends with post-consumed r-PET (bottle flakes wastes) were prepared with different compositions using a twin screw extruder (TSE) while CE was also incorporated to further improve the properties of the noted blends. Injection molding was then employed to prepare the testing samples. The melt behavior, miscibility, phase separation, crystallization behavior, solid viscoelastic features, and mechanical properties of the blends are comprehensively discussed in this study. More specifically, we illustrated how the miscibility and phase separation of PET and PBT amorphous and crystalline regions are dependent on the cooling rate of the blends, or in other word, on the cooling occurs in processing of the blends. It has been elucidated that the PET and PBT crystalline phases could be immiscible when the samples are quickly cooled. However, this phase immiscibility of the crystalline phases could turn to be phase miscibility and co-crystallization of PET and PBT could occur when the cooling rate decreases. In this context, the miscibility and phase separation behaviors of the crystalline phases of PET and PBT and their dependency on the cooling gradients are disclosed in this study more comprehensively.
Experimental
Materials
A commercial PBT with 17 ± 4 cm3/10 min melt volume rate was supplied from Sasa Polyester Inc. Co. in Adana, Turkey. R-PET flakes obtained from bottle wastes were supplied from Çevre PET Inc. Co. in Adana/Turkey. The properties and contamination contents of supplied r-PET flakes are listed in Table 1. Thermal degradation temperatures and residual concentrations of bottle-grade virgin PET, processed r-PET were determined by using TA Instrument thermogravimetric analyzer (TGA Q 500). Analysis were carried out at a heating rate of 20 °C/min from room temperature to 950 °C. The percentage of the residues at 600 °C were recorded in both water bottle grade prime PET and processed r-PET. Similar amounts of inorganic residues (11.2 wt% and 11.7 wt%) were observed in prime PET and processed r-PET, respectively, as also stated in Table 1.
A commercial CE with Joncryl ADR 4468® trade name was supplied from BASF. Joncryl is a multi-functional styrene acrylic oligomer which includes epoxy reactive groups with a chemical structure which is reported elsewhere [41].
Processing and Sample Preparation
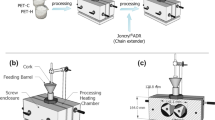
PBT pellets and r-PET flakes were dried in a vacuum oven at 70 °C overnight to remove moisture and avoid further degradation during processing. A laboratory scale TSE with 24 mm screw diameter and 28 L/D ratio was used for preparing the compounds. The temperature profile was set to 235–245–255–260–260–255–245 °C from the feeder to the die with a constant screw rotation speed of 200 rpm. After drying, the samples were taken out from the oven immediately and processed right away to minimize the humidity absorption. The filament shaped extrudate were cooled in a water bath and then cut into pellets in a granulating unit and dried in the oven right away. The experiments were designed and conducted as follows:
-
(a)
Various contents of CE, Joncryl, (0, 0.05, 0.25, 0.40, and 0.80 wt%) were, firstly, melt blended with r-PET flakes using the TSE to obtain chain extended r-PET. The PET and the CE were separately added into the extruder through two gravimetric hoppers at the noted barrel temperature profile which is appropriate for the CE reaction [41]. To keep the same thermal history, r-PET samples were also processed under similar conditions. Codifications and compounding ratios of the noted CE loadings within r-PET flakes are summarized as: r-PET processed, r-PET + 0.05CE, r-PET + 0.25CE, r-PET + 0.40CE, and r-PET + 0.80CE, respectively.
-
(b)
The r-PET samples were then melt blended with PBT via TSE. The r-PET ratios were fixed at 25, 50 and 75 wt%. Codifications of the neat PBT/r-PET blends are also summarized as: 75PBT/25r-PET, 50PBT/50r-PET, and 25PBT/75r-PET. CE was also separately melt mixed with similar PBT/r-PET blend ratios with fix CE content of 0.2 wt%. The compounding ratios and the codifications of these blends are also referred to as: 75PBT/25r-PET/0.2CE, 50PBT/50r-PET/0.2CE, and 25PBT/75r-PET/0.2CE. In blend of 50PBT/50r-PET, the CE content of 0.4 wt% was also explored separately to further understand the CE content effect.
-
(c)
After drying the pelletized extrudate in a vacuum oven at 70 °C overnight, injection molding machine Arburg Allrounder with 25 mm screw diameter and 24 L/D ratio was used to prepare the standard DMA, tensile and impact test samples. The barrel temperature was set to 260–265–265–270–275 °C from the hopper to the nozzle. The mold was controlled at room temperature and injection pressure was set to 1500 bar. The r-PET and its chain extended samples prepared in section (a) could not be processed through the injection molding due to its poor melt features and hence the DMA and mechanical properties of these samples could not be measured.
Melt Flow Index (MFI)
Melt flow rate (MFR) values of the samples were obtained from MFI test according to ISO 1133 standard. The temperature and the stable weight were set to 260 °C and 2.16 kg, respectively. For each formulation, the weight of three specimens was measured and recorded. The average value of the three results was then reported with g/10 min unit.
Differential Scanning Calorimetry (DSC)
The melting and cooling related thermal studies of the samples were performed using TA-Instrument DSC Q 200 under 50 ml/min flow rate of inert nitrogen gas. Thermal analysis of samples was performed by heating the samples from 30 to 300 °C followed by cooling to 30 °C and reheating to 300 °C during a second scan. Further studies on crystallization behavior of the samples were examined through DSC at different cooling rates of 2, 5, 10 and 20 °C/min followed by reheating to 300 °C with constant heating rate of 5 °C/min.
Dynamic Mechanical Analysis (DMA)
Temperature sweep experiments were applied to specimens through dynamic mechanical analysis (DMA) by using TA Instrument DMA Q 800 under dry air in dual cantilever mode. According to ASTM D7028-7, the rectangular standard test specimens’ dimensions were thickness of 3.18 ± 0.2 mm, width of 12.80 ± 0.2 mm and length of 59.75 ± 0.2 mm. Heating rate was set to 2 °C/min from 30 to 130 °C at a constant frequency of 1 Hz, 0.03% strain and 15 µm amplitude.
Mechanical Properties
Tensile properties of dog bone shaped specimens were measured at room temperature by using a tensile testing machine, Zwick Z 020, according to ASTM D638 M-91a test standard. The cross-head speed was set to 50 mm/min and gauge length (Lo) was set to 70 mm. The average value of the five specimens is reported in this study.
The Izod impact strength of the standard samples was measured using a standard pendulum type hammer mounted on a machine (Zwick Roell HIT 5.5P), according to ISO 180 standard. The dimensions of rectangular test specimens were 4 ± 0.2 mm, 10 ± 0.2 mm, and 80 ± 0.2 mm. Standard 2 mm V notch was also generated in the impact samples by using a manual notch cutter machine. The notched surfaced of the specimens faced to the pendulum in the test machine. The average value of ten results is reported in this study.
Results and Discussion
Melt Flow Behavior
MFR values of the samples were measured to estimate their melt behavior. Enhanced melt strength could result in decrease in MFR values. It should also be noted that the processing could separately reduce the melt strength via shear thinning and/or mechanical and thermal degradation of the samples and hence these could further reduce the processability. MFR values of PBT and r-PET before and after processing are given in Fig. 1a. PBT showed higher melt strength than r-PET with lower MFR value. MFR of PBT increased around 14% after processing however, the MFR of r-PET increased over 60% after processing. Higher increase in MFR of r-PET after processing shows its sensitivity to processing due to its high potential for thermal degradation and hydrolysis.

MFR values of the virgin and processed PBT and r-PET (a), r-PET samples with CE (b) and blends without CE (c) and with CE (d)
Branching r-PET with various concentrations of CE enhanced the melt strength and decreased the MFR values as shown in Fig. 1b. The addition of 0.25 wt% of CE almost compensated the melt strength loss of r-PET after processing. The lowest MFR value of 27 g/10 min was achieved by addition of 0.8 wt% CE into r-PET. Increase in melt strength of branched r-PET could associate with molecular weight increase due to branching during extrusion, as MFR strongly depends on molecular weight.
In blends of PBT/r-PET, increasing the amount of r-PET resulted in higher MFR values, due to the higher MFR value of r-PET itself (Fig. 1c). However, blending r-PET with PBT could still compensate the melt strength loss of r-PET after processing to some extent. The lowest MFR value of 22 g/10 min was achieved in 75PBT/25r-PET blend. Comparing the MFR of the neat blend samples with the blends containing CE (Fig. 1d), it is shown that the addition of 0.2 wt% CE decreased the MFR values of the blends in all blending ratios, however, this decrease was more significant in 50PBT/50r-PET blend. It was further observed that in 50PBT/50r-PET blend the increase in CE content to 0.4 wt% further decreased the MFR value from 22 to 14 g/10 min (not shown here).
Thermal and Crystallization Behavior
Branching r-PET with CE
First heating, cooling and second heating curves of processed PBT and processed r-PET samples are shown in Fig. 2. While Tg of PBT is around 58 °C, that of r-PET is ~ 79 °C. During the first heating cycle, a cold crystallization peak of r-PET was observed around 121 °C. The total crystallinity of the processed samples were calculated using the enthalpy required to melt 100% crystalline structure of PET and PBT which are 140 and 145 J/g, respectively [42,43,44]. After fast cooling subsequent to processing, the processed r-PET and PBT showed 11 and 35% crystallinity, respectively. Due to the incomplete crystallization of r-PET, cold crystallization was also observed at 121 °C. The crystal melting temperatures of processed r-PET and PBT were revealed as 249 and 224 °C, respectively. During the cooling cycles at the slow rate of 5 °C/min, PBT revealed earlier and faster crystallization with total crystallinity of 39% whereas r-PET showed slower crystallization but as the consequence of slow cooling 30% crystallinity could be formed. During the second heating cycle, both polymers showed double melting peak which could be attributed to presence of crystals with different perfections when slowly cooled.

First heating, cooling and second heating thermograms of processed PBT and r-PET
In order to investigate how the cooling rate could influence the degree of perfection of the crystals and hence the structure of the crystal melting peaks, the samples were cooled at different rates and their second heating thermograms were compared. Figure 3 shows the crystallization (initiation and peak) temperatures of processed PBT and r-PET during cooling cycles at different cooling rates. Figure 4 also illustrated how various cooling rates could affect the second heating thermograms of the processed PBT and r-PET. The first heating of the processed samples is also included in the graphs which passed through a very fast cooling, where is referred to as quenched samples.

Crystallization (initiation and peak) temperatures of processed PBT and r-PET during cooling cycles at different cooling rates

Second heating thermograms subsequent to different cooling rates of processed PBT and r-PET using DSC (Pink graphs are the first heating graphs of the extruded samples)
As shown in Fig. 3, under different cooling rates, the difference between the crystallization initiation and peak temperatures is almost constant in PBT. This means that the cooling rate could not hinder the fast crystallization of PBT. However, in r-PET, as the cooling rate increases, this temperature difference increases due to the reduced crystallization rate. In Fig. 4, it is clear that for r-PET when the cooling rate increases too much, the crystallization becomes very slow and the crystallinity decreases whereas the crystallinity in PBT, even after the processing, is still very high (~ 35%). On the other hand, after cooling at slow rates, crystallization occurred at higher temperatures which should have caused the formation of more closed-packed crystals. However, during the second heating (Fig. 4), the melting behavior of the slowly cooled samples appeared with a more obvious peak/shoulder at lower temperatures. As the cooling rate increased, the low melting peak/shoulder became smaller and eventually rapidly cooled samples revealed single peaks at higher temperatures. This could be due to the higher chance of transesterification during the slow cooling rates where it could cause formation of less closed-packed crystals although the crystallization has occured at higher temperatures with high molecular mobility. This phenomenon seems to be valid for both PBT and r-PET which could encounter transesterification at high temperatures.
The crystallization behavior of the processed r-PET samples melt blended with CE was also explored during the first heating thermograms and along various cooling cycles to explore the effect of branching on PET’s crystallization behavior. Figure 5a reveals the first heating thermograms of the r-PET samples with different CE contents and Fig. 5b shows the crystallization rate during the cold crystallization. As shown, the addition of CE up to 0.25 wt% expedites the crystallization of r-PET, most probably due to the role of branched sites as nucleation regions [22, 23], however beyond this value specially at 0.8 wt% CE content the crystallization could be suppressed due to the reduced molecular mobility. The cooling graphs of the r-PET branched samples are also shown in Fig. 6. The addition of CE up to 0.4 wt% could expedite the crystallization rate and and degree of crystallinity of r-PET. This, again, could have been caused by the action of branched molecular structures as nucleating points which promoted the crystallization [22, 23]. On the other hand, with the addition of 0.8 wt% CE (highly branched r-PET), both the crystallization rate and degree of crystallinity of PET were hindered due to the lower mobility of branched molecules. Moreover, the addition of CE did not affect the glass transition and melting temperatures at the first and second heating graphs.

First heating thermograms of r-PET samples with various CE contents (a) and the relative cold crystallization of the noted samples versus time (b)

Cooling thermograms of the r-PET samples with various CE contents
Binary Blends of PBT/r-PET: Phase Miscibility
Figure 7 shows the heat/cool/heat thermal behavior of PBT/r-PET blends under heating and cooling rates of 5 °C/min. During the first heat, which reflects the thermal behavior of the rapidly cooled processed samples, two separate melting peaks of PBT and r-PET crystals are clearly appeared whereas single Tg values exist. This reveals the full miscibility of both polymers in amorphous phases and immiscibility within the crystalline phases. During the fast cooling, PBT crystallizes very rapidly at higher temperatures whereas the crystallization of PET occurs at lower temperatures. Thereby two different crystallization sequences occur at different occasions. It is interesting to see that in the blend systems, the r-PET samples showed much slower cold crystallization. This is probably because the PBT crystallites could enhance the PET’s crystallization rate and causes heterogeneous nucleation of PET crystals at lower temperatures. Therefore, blending PET with PBT could improve the crystallization kinetics of PET where PBT crystals could behave as crystal nucleating agents. When the samples were cooled very slowly (Fig. 7b), the crystallization of the whole blend occurred more slowly and at lower temperatures. In 50PBT/50r-PET samples the crystallization temperature reduced to almost around 160 °C. Figure 7c also reveals the heating thermograms of the slowly cooled samples. It is clear that the melting peaks are not only appeared at much lower temperatures but also the separate melting peaks that appeared in the first heating are merged and almost unified in a single wider peak. As Fig. 3 showed, during the slow cooling rates, the crystallization of PBT and r-PET almost occur simultaneously. Therefore, during slow cooling rates, the chance of co-crystallization and maybe transesterification increase and hence low perfected crystals containing both PBT and r-PET molecules could form. Figure 8 also reveals the heating thermograms of the blend samples subsequent to various cooling rates. This could clearly be seen that when the cooling rate decreases the chance of co-crystallization with smaller and/or less perfect crystals increases. With the decrease in cooling rate, the melting peaks with more unified peak structure appeared at lower temperatures. Therefore, when the blends are slowly cooled the full miscibility of both amorphous and crystalline phases could be observed. Whereas fast cooling results in immiscibility of crystalline phases and the earlier crystallization of PBT enhances the crystal nucleation power of PET and hence the PET’s crystallization could also be improved. As shown, the amount of crystallinity of the blends does not change dramatically with the cooling rate.

First heating (a), cooling (b) and second heating (c) thermograms of binary blends of PBT/r-PET

Second heating thermograms of PBT/r-PET blends subsequent to different cooling rates: a 2, b 5, c 10, and d 20 °C/min
The DSC thermal behavior of PBT/r-PET blends were also compared with and without the presence of 0.2 wt% of CE. The results revealed that the thermal behavior of 75PBT/25r-PET and 25PBT/75r-r-PET blends does not change dramatically when also melt mixed with 0.2 wt% CE (not shown here), however in the 50PBT/50r-PET blends, the differences could be more evident. In order to understand the CE effect in the noted blend, 0.4 wt% of CE was also melt blended separately. Figure 9 shows the crystallization initiation and peak temperatures of 50PBT/50r-PET blends with 0, 0.2, and 0.4 wt% of CE as a function of cooling rate. Figure 10 also reveals how the heating thermograms subsequent to various cooling cycles could look like. As also observed in Fig. 3, generally with the increase of cooling rate the crystallization tends to appear at lower temperatures due to the reduced diffusion time and crystallization kinetics. In Fig. 9, in all blends specially in the case without CE, the crystallization temperature decreased when the cooling rate increased from 2 to 5 °C/min. It seems that the co-crystallization occurs at both 2 and 5 °C/min, but due to the increased cooling rate the crystallization occurred at lower temperature when cooled at 5 °C/min. However, beyond 5 °C/min the crystallization tends to occur earlier as the co-crystallization might not tend to occur and even the increased cooling rate doesn’t cause crystallization at lower temperatures. Thereby, this increase in crystallization temperature with increased cooling rate could be due to the increased chance of separate crystallization of PBT and r-PET at earlier temperatures with more closed packed structure. In other words, similar to what was shown earlier, the chance of crystal phase immiscibility could increase with cooling rate. This separate crystallization of PBT and r-PET was more obvious in blends with no CE. It should also be noted that the rate of crystallization may be considered constant during all cooling rates by considering the constant temperature differences between crystal initiation and peak points. In blends with CE, these behaviors were less evident. In presence of CE, during slow cooling, more sever co-crystallization could occur with more merged structure as the CE could bring the molecules of both PBT and r-PET more along each other to form ordered structure. During cooling at the rate of 2 °C/min, almost one melting peak could be observed with less ordered structure. This becomes more obvious when the CE content increased. During fast cooling, it seems that the separate crystallization could occur and this become again more evident with increased amount of CE.

Crystallization (initiation and peak) temperatures of 50PBT/50r-PET blends with and without CE during cooling cycles at different rates

Second heating thermograms subsequent to different cooling rates of 50PBT/50r-PET blends without CE (a), with 0.2 wt% (b) and with 0.4 wt% (c) CE
Solid Viscoelastic Behavior
Storage modulus and tan delta values versus temperature were recorded for various blend samples and are shown in Fig. 11. Figure 12a also reveals the storage modulus values of blend samples at 30 and 80 °C. The Tg values recorded from the tan delta graphs are also reported in Fig. 12b. Addition of r-PET into PBT slightly decreased storage modulus at room temperature. This decrease was slightly more evident in blends with CE most probably due the existence of less closed packed co-crystals and miscible crystalline phase. All blends showed lower storage modulus at rubbery state and the addition of r-PET and CE into PBT decreased the storage modulus more significantly within this region. This is because beyond the Tg, the rigidity of the samples derives from mainly the crystalline phase. Therefore, existence of less close packed co-crystals could more dominantly show its negative effect on reduction of storage modulus. The increase in modulus in the rubbery region caused by the cold crystallization is more evident in the 20PBT/75r-PET blends due to existence of more PET content. The single Tg of the blends also increased with the increase in r-PET and further decreased in presence of CE which might be due to the more active transesterification in presence of CE. Processed PBT possessed the lowest tan delta with lower energy dissipation, while 20PBT/75r-PET blend had the highest which reflects the lower impact behavior with the increase in PET content.

Storage modulus (a, c) and tan delta (b, d) graphs of blends during the temperature sweep experiments

Storage modulus at 30 and 80 °C (a) and Tg (b) values of the blends samples
Mechanical Properties
Young’s modulus (E) and tensile strength values of the blends were recorded from tensile test. Figure 13 reveals the mechanical properties of the blends during tensile and impact testing. The results showed that when r-PET is blended with PBT, the modulus and strength do not differ much from the processed PBT. The addition of CE also did not show significant effect on changing the properties although it significantly influenced the processing of the blends due to the increased melt strength. The increase in PET content also increased the ductility of PBT and this becomes more evident when CE was also melt blended with PBT/r-PET. From the impact test, it is shown that the impact strength of the blends reduced from that of processed PBT and the addition of r-PET decreased impact strength of PBT. This could also be predicted from the tan delta values reported in Fig. 11b and d.

Strength and modulus (a), strain at break (b) of the blend samples during tensile testing and impact strength of the samples during Izod impact experiments (c)
Conclusion
In this study, PBT/r-PET binary blends were developed at different ratios. CE with a trade name of Joncryl ADR 4468 was also used to be melt mixed with the noted blends at a fix content. For this purpose, the r-PET was also melt blended with CE and its melt and thermal behavior were explored. The CE decreased the r-PET’s MFR consistently. The use of CE up to 0.4 wt% also increased the crystallinity and crystallization rate of r-PET. At low contents, CE could act as nucleating agent and promote crystallization. On the other hand, 0.8 wt% CE hindered the crystallization of r-PET due to reduced chain mobility.
In the case of PBT/r-PET blends, increased amount of r-PET could decrease the amount of crystallinity. All blends either with or without CE showed single Tg. PBT/r-PET blends showed miscibility at amorphous state and immiscibility in crystalline phases. After slow cooling rates, however, individual crystal melting peaks of PBT and r-PET in the first heating cycle turned into single peak with some shoulders. When the samples were cooled slowly the chance of co-crystallization increased and hence PBT/r-PET blends also showed miscibility at crystal melting state. In other words, as the process cooling increases the crystallization could occur separately with immiscible crystalline phase structure and when the cooling rate decreases co-crystals with less closed packed structure and lower melting peaks could be formed as a miscible phase.
Addition of r-PET decreased storage modulus and impact behavior during the temperature sweep test. Addition of CE also decreased the storage modulus due to the existence of less perfected co-crystals within the rubbery region. CE, on the other hand, increased the melt strength and hence processability in extrusion process by branching molecules of r-PET and PBT/r-PET blends. The modulus and strength during tensile testing of the blends were pretty much similar to that of PBT, whereas the ductility increased with PET. On the other hand, the impact strength of blends was lower than processed PBT.
References
Utracki LA (2013) Commercial polymer blends. Springer, Dordrecht
Kannan G, Grieshaber SE, Zhao W (2016) Thermoplastic polyesters. In: Olabisi O, Adewale K (eds) Handbook of thermoplastics. CRC Press, pp 319–347
Lepoittevin B, Roger P (2011) Poly(ethylene terephthalate). In: Thomas S, Visakh PM (eds) Handbook of engineering and speciality thermoplastics. Wiley, Scrivener, pp 97–125
Awaja F, Daver F, Kosior E, Cser F (2004) The effect of chain extension on the thermal behaviour and crystallinity of reactive extruded r-PET. J Therm Anal Calorim 78:865–884
Galanty PG, Richardson JJ (1988) Polyethylene terephthalates (PET). In: Engineering plastics. Engineered materials handbook, vol 2. ASM International, Metals Park, pp 172-176
Al-Sabagh AM, Yehia FZ, Eshaq G, Rabie AM, ElMetwally AE (2016) Greener routes for recycling of polyethylene terephthalate. Egypt J Pet 25:53–64
Gourmelon G (2015) Global plastic production rises, recycling lags. New Worldwatch Institute analysis explores trends in global plastic consumption and recycling. Retrived from http://vitalsigns.worldwatch.org/sites/default/files/vital_signs_trend_plastic_full_pdf.pdf
Arena U, Mastellone ML, Perugini F (2003) Life cycle assessment of a plastic packaging recycling system. Int J Life Cycle Assess 8:92–98
Karsli NG (2015) A study on the fracture, mechanical and thermal properties of chain extended recycled poly (ethylene terephthalate). J Thermoplast Composite Mater 30(8):1157–1172
Nofar M, Salehiyan R, Ray SS (2019) Rheology of poly (lactic acid)-based systems. Polym Rev 3:3. https://doi.org/10.1080/15583724.2019.1572185
Nofar M, Park CB (2014) Poly (lactic acid) foaming. Prog Polym Sci 39(10):1721–1741
Rieckmann T, Völker S (2004) Poly (ethylene terephthalate) polymerization–mechanism, catalysis, kinetics, mass transfer and reactor design. In: Scheirs J, Long TE (eds) Modern polyesters: chemistry and technology of polyesters and copolyesters. Wiley, New York, pp 29–115
Awaja F, Daver F, Kosior E (2004) Recycled poly (ethylene terephthalate) chain extension by a reactive extrusion process. Polym Eng Sci 44:1579–1587
Incarnato L, Scarfato P, Di Maio L, Acierno D (2000) Structure and rheology of r-PET modified by reactive extrusion. Polymer 41:6825–6831
Daver F, Gupta R, Kosior E (2008) Rheological characterisation of recycled poly (ethylene terephthalate) modified by reactive extrusion. J Mater Process Technol 204:397–402
Japon S, Boogh L, Leterrier Y, Månson JA (2000) Reactive processing of poly (ethylene terephthalate) modified with multifunctional epoxy-based additives. Polymer 41:5809–5818
Bikiaris DN, Karayannidis GP (1996) Thermomechanical analysis of chain-extended PET and PBT. J Appl Polym Sci 60:55–61
Makkam S, Harnnarongchai W (2014) Rheological and mechanical properties of r-PET modified by reactive extrusion. Energy Procedia 56:547–553
Raffa P, Coltelli MB, Savi S, Bianchi S, Castelvetro V (2012) Chain extension and branching of poly (ethylene terephthalate) (PET) with di-and multifunctional epoxy or isocyanate additives: an experimental and modelling study. React Funct Polym 72:50–60
Rosu RF, Shanks RA, Bhattacharya SN (1999) Shear rheology and thermal properties of linear and branched poly (ethylene terephthalate) blends. Polymer 40:5891–5898
Li G, Yang SL, Jiang JM, Wu CX (2005) Crystallization characteristics of weakly branched poly (ethylene terephthalate). Polymer 46:11142–11148
Nofar M, Zhu W, Park CB, Randall J (2011) Crystallization kinetics of linear and long-chain-branched polylactide. Ind Eng Chem Res 50:13789–13798
Nofar M (2018) Synergistic effects of chain extender and nanoclay on the crystallization behavior of polylactide. Int J Mater Sci Res 1(1):1–8. https://doi.org/10.18689/ijmsr-1000101
Nofar M, Sacligil D, Carreau PJ, Kamal MR, Heuzey MC (2019) Poly (lactic acid) blends: processing, properties and applications. Int J Biol Macromol 125:307–360
Utracki LA, Mukhopadhyay P, Gupta RK (2014) Polymer blends: introduction. Polymer blends handbook. Springer, Berlin, pp 3–170
Barlow JW, Paul DR (1981) Polymer blends and alloys—a review of selected considerations. Polym Eng Sci 21:985–996
Escala A, Stein RS (1979) Crystallization studies of blends of polyethylene terephthalate and polybutylene terephthalate. Polym Eng Sci 37:91–95
Avramova N (1995) Amorphous poly (ethylene terephthalate)/poly (butylene terephthalate) blends: miscibility and properties. Polymer 36:801–808
Ito K, Haraguchi Y, Hayakawa S, Toda A (2008) Enhanced crystallization of blended poly (ethylene terephthalate) and poly (butylene terephthalate). Polym J 40:992
Zhang Z, Feng L, Li Y, Wang Y, Yan C (2015) Nonisothermal crystallization kinetics of poly (butylene terephthalate)/poly (ethylene terephthalate)/glass fiber composites. Polym Compos 36:510–516
Szostak M (2004) Mechanical and thermal properties of PET/PBT blends. Mol Cryst Liq Cryst 416:209–215
Rajakumar PR, Nanthini R (2011) Thermal and morphological behaviours of polybutylene terephthalate/polyethylene terephthalate blend nanocomposites. Rasayan J Chem 4:567–579
Akkapeddi MK (2003) Commercial polymer blends. Polymer blends handbook. Springer, Dordrecht, pp 1023–1115
Aravinthan G, Kale DD (2005) Blends of poly (ethylene terephthalate) and poly (butylene terephthalate). J Appl Polym Sci 98:75–82
Tao J, Jin HF, Sun T (1991) A study on compatibility of PBT/PET binary blended fibre. Plast Rubber Composites Process Appl UK 16:49–53
Lang X, Dong X, Jiyong M, Jing L, Lijuan W (2015) CN Patent No. 102911486 (B). Retrived 13 April 2017
Matthias B, Ulrich P (2014) U.S. Patent No. 2014163156 (A1). Retrieved 13 April 2017
Jin JH, Dae CG, Chul CH, Hyun KS (2013) KR Patent No. 20130021502 (A). Retrieved 13 April 2017
Baxi RN, Pathak SU, Peshwe DR (2010) Mechanical, thermal, and structural characterization of poly(ethylene terephthalate) and poly(butylene terephthalate) blend systems by the addition of postconsumer poly(ethylene terephthalate). J Appl Polym Sci 115:928–934
Rahmat AR, Lim PS, Sin LT, Bee ST, Tee TT (2012) The effects of CE on viscosity and mechanical properties of poly (buthylene terephthalate) blending with recycled poly (ethylene terephthalate)-glass fiber composite. J Appl Sci 12:296–300
Villalobos M, Awojulu A, Greeley T, Turco G, Deeter G (2006) Oligomeric CEs for economic reprocessing and recycling of condensation plastics. Energy 31:3227–3234
Cheng SZD, Pan R, Wunderlich B (1988) Thermal analysis of poly(butylene terephthalate) for heat capacity, rigid amorphous content, and transition behavior. Macromol Chem Phys 189:2443–2458
Wunderlich B (1980) Macromolecular physics, vol 3. Crystal melting. Academic Press, New York
Pospiech D, Häußler L, Korwitz A, Fischer O, Starke S, Jehnichen D, Koppl T, Altstadt V (2012) The miscibility of poly(butyleneterephthalate) (PBT) with phosphorus polyester flame retardants. High Perform Polym 24(1):64–73
Acknowledgment
The authors would like to thank to Arcelik A.S. and Central Research and Development Director Cem Kural, Materials Technologies Research and Development Department Manager Dr. Mustafa Sezer, Polymer & Chemistry Team Leader Dr. Yusuf Yusufoğlu, Research and Development Specialist Ceren Kovanci, Research and Development Technicians Metehan Cihangir and Sefa Yasin Uzen for supporting the research activities conducted in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nofar, M., Oğuz, H. Development of PBT/Recycled-PET Blends and the Influence of Using Chain Extender. J Polym Environ 27, 1404–1417 (2019). https://doi.org/10.1007/s10924-019-01435-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-019-01435-w