
Abstract
This article is a mini-review mostly based on the work of the authors’ laboratory on the redox chemistry of metallodendrimers and gold nanoparticles, with emphasis on “click” chemistry. Late transition-metal sandwich complexes possess a rather unique ability to withstand two or three oxidation states without breakdown, especially with permethylated π-cyclopentadienyl or arene ligands. When they are linked to dendritic cores, the assembled nano-systems undergo chemically and electrochemically reversible transfer of a large number of electrons (up to 14,000). These multiple redox processes are useful for nanodevices behaving as nanobatteries for redox sensing, modified electrode surfaces and redox catalysis. Click chemistry was recently disclosed as one of the most powerful means to form such assemblies including both arene-cored and gold nanoparticle-cored dendrimers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The inorganic chemistry of metals offers a variety of materials properties [1], and for instance solid state materials based on lithium composites presently dominate the market of batteries [2]. Redox and electron-transfer properties are also a great richness of transition-metal compounds related to their multiple redox states that are reflected in key biological functions [3]. Therefore molecular transition-metal chemistry can benefit from the lessons of both the solid state and biological areas to design new aspects of redox properties and functions [4] that should prove of interest in nanoscience [5].
A few families of molecular transition-metal complexes present a robustness of two redox states, which may be useful for the design of nanodevices involving reversible electron-transfer processes. They include metalloporphyrins and related metalla-macrocycles [6], some large inorganic clusters [7] and first-raw late transition-metal sandwich complexes, typically with iron and cobalt that present the advantage of biocompatibility [8]. In this article, we review chemistry mostly originated from the authors’ laboratory on the design, characterization and redox functions of molecular, dendritic and nanoparticle materials based on these iron and cobalt sandwich complexes, and in particular we survey the contribution of click chemistry to this field.
2 Electron-Transfer Reactions of Redox-Active Organometallic Reagents
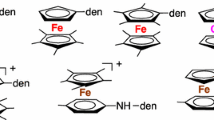
Electron-transfer reactions and processes of inorganic and organometallic complexes play important roles in molecular chemistry [4, 9], in particular for redox reagents, [10–14] sensors [15–19], catalysts [4, 9, 20–23] and as parts of photochemical devices [4, 9]. Since the discovery of the 18-electron sandwich structure of ferrocene [8], Wilkinson et al. [24] noted its reversible oxidation to ferricinium at modest potentials, and the isolation of ferricinium salts provided useful mild single-electron oxidants. Subsequently, the organometallic community has frequently used ferricinium as an oxidant [10–14] and also sometimes cobaltocene [25–27] as reductant [10–14]. Various transition-metal sandwich complexes have been selected as a library of redox reagents and optimized for organometallic single electron-transfer reactions [13, 14] (see for instance Fig. 1). Another useful application has been the utilization of permethylated metallocenes as references for the determination of electrochemical standard redox potentials, because contrary to parent metallocenes the encapsulated metal centers therein are well protected against nucleophilic interactions with solvent molecules and anions, which makes the redox potential independent of the nature of solvents and electrolytes [28, 29].

Late transition-metal sandwich complexes as organometallic redox reagents
3 Electron-Reservoir Transition-Metal Sandwich Complexes
Some time ago, mono- [30] and bimetallic [31, 32] iron-sandwich complexes of the [Fe(η5-Cp)(η6-arene)] family (Cp = η5 cyclopentadienyl) were found to be particularly suitable for such reversible electron-transfer processes, and were therefore designated as electron-reservoir systems, also because the redox potentials of the 18/19-electron interchange was located at a very negative potential values, which meant that the 19-electron complexes were very electron-rich and behaved as strong reductants [33] (Fig. 2). Indeed, these complexes, such as the prototype 19-electron complex [FeI(η5-Cp)(η6-C6Me6], were found to reduce oxygen to superoxide radical anion, C60 to its mono-, bis- and tri-anions, and a variety of substrates including CO2 and nitrate and nitrite anions [8]. With these later anions, the 19-electron complexes were shown to be redox catalysts for their cathodic reduction to ammonia in water [34]. Not only were these compounds redox catalysts, but they also disclosed the properties of electrocatalysts, i.e. catalysts of electron-transfer-chain reactions such as ligand-substitution processes for a variety of robust 18-electron organometallic complexes [35–37]. The 19-electron complexes [FeI(η5-C5R5)(η6-C6Me6], R = H or Me, are in fact so electron-rich that their ionization potentials determined by He(I) photoelectron spectroscopy were found to be of the same order as that of potassium metal, which made these complexes the most electron-rich neutral compounds known even up to now [38]. The 20-electron complex [Fe(η6-C6Me6)2] also undergoes remarkable and synthetically useful electron-transfer reactions, although it is less electron-rich than [FeI(η5-Cp)(η6-C6Me6] [39].

19-electron complexes of the family [FeI(η5-C5R5)(η6-C6R’6]: Complex 1 is not thermally stable and dimerizes at 0 °C. Complex 2 is the prototype of electron-reservoir 19-electron complexes. It is thermally stable up to 100 °C. Complexes 3 are known for a variety of R groups including dendritic ones. Complex 4 is known as one of the most electron-rich neutral compounds (together with [FeI(η5-C5Me5)(η6-C6Et5H]). For E 0 values, see Fig. 1 [30, 38]
4 Reservoirs of Electron Holes: Strong Oxidants
Interestingly, the same series of complexes, [Fe(η5-C5R5)(η6-C6Me6)][PF6], for which oxidation was originally believed to be impossible as they were stable in concentrated sulfuric acid [40, 41], could be reversibly oxidized at very positive potentials in SO2 or MeCN using SbF5 or SbCl5 and the 17-electron complex [FeIII(η5-C5Me5)(η6-C6Me6)][PF6][SbCl6] (Fig. 1) could even be isolated and serve as very strong oxidant and redox catalyst for anodic oxidation reactions [42]. This chemistry demonstrated the possibilities of isolation and full characterization of very well-defined 17- to 19-electron complexes of the same family and disclose a large spectrum of useful redox properties and functions as stoichiometric and catalytic reagents for redox reactions in organic, inorganic and materials chemistry that are still in use to-day (Scheme 1).

Syntheses of the deep-purple 17-electron and dark-green 19-electron complexes of the family [Fe(η5-C5R5)(η6-C6Me6)]+2/0 (R = H or Me) from the yellow 18-electron complexes. Both the 17-electron and 19-electron complexes are thermally stable for R = Me (Color figure online)
5 Dendritic Ferrocene, Cobaltocene and [Fe(Cp)(η6-arene)] Complexes
The dendritic structures offer the opportunity of introduction of many redox centers at the periphery of the molecular frame [43–46], and this concept has been largely developed with ferrocene derivatives given their rich chemistry allowing multiple possibilities of functionalization [46–49]. Dendritic ferrocenes can be used as oxo-anion redox sensors with positive dendritic effects, i.e. simple mononuclear amidoferrocenes show interactions with anions that are too weak to be electrochemically detectable, but the variation of amidoferrocenyl redox potentials upon addition of oxoanions becomes all the larger as the dendrimer generation increases [50]. The amide junction is useful both for ferrocene functionalization onto dendrimers [47, 50] and for providing H-bonding with oxo-anions that contributes to their redox recognition [50]. Redox anion recognition indeed relies on the synergy between the electrostatic, supramolecular (H-bonding) and topological factors, among which the latter is not the least one [15, 19]. For instance, even if the supramolecular (H-bonding) factor sometimes plays a major role, it has been shown that redox recognition was possible in its absence with giant dendritic alkylferrocenes [51]. Such large ferrocenyl dendrimers were all the more easily accessible as the dendritic construction involved a 1 → 3 connectivity that led to a much faster increase of the number of terminal tethers than the more classic 1 → 2 connectivity encountered in most dendrimers [45, 52]. The 1 → 3 connectivity, pioneered by Newkome for dendrimer construction [45, 53], is also accessible upon connecting dendritic core with tri-branched ferrocenyl dendrons using silicon-based links available by catalyzed hydrosilylation reactions [47, 48], Williamson reactions with phenolates [48] and even upon selective cross olefin metathesis between olefin-terminated dendrimers and dendronic acrylates [54]. Large ferrocenyl dendrimers terminated with up to 14,000 ferrocenyl units at the periphery show chemically and electrochemically reversible cyclic voltammetry waves indicating fast electron-transfer between all the redox sites and the electrode (Figs. 3, 4).


AFM of arene-cored G4 (theoretically 729 ferrocenyl termini; calc. 700 ± 40; height: 3.5 ± 0.3 nm) to G7 metallodendrimers (theoretically 19,683 ferrocenyl termini; calc.: 14,000 ± 1,000; height: 7.5 ± 1.5 nm) deposited from CH2Cl2 solutions on mica surfaces [49]. See the molecular structure of G2 in Fig. 3
The AFM show packages of dendrimers that make particles of remarkably homogeneous size, this homogeneity culminating for generation 4 with a theoretical number of 729 ferrocenyl groups (Fig. 4). Thus it appears that the aggregation of ferrocenyl dendrimers into nanoparticles on surfaces occurs in the same fashion as aggregation of metal atoms in weakly dispersed metal nanoparticles.
Ferrocenyl dendrimers have in fact been synthesized with a large variety of cores from organic to inorganic (Mo6 clusters; gold nanoparticles, vide infra) and even with silica nanoparticle cores [55]. It was also possible to synthesize large cobalticinium dendrimers with the same organic dendrimer cores (Fig. 3), although the largest cobalticinium dendrimers showed broad cyclic voltammograms possibly due to some electrostatic differentiation [56]. Finally linkage between amino-terminated dendritic cores and carboxylic acid derivatives of CpFe(η6-arene) salts in which the substituent is located on the Cp ligand (i.e. 3 in Fig. 2) provided either metallodendrimers as ammonium carboxylate salts or covalent amides upon reaction with the chlorocarbonyl derivatives. In the case of [Fe(η5-C5H5COCl)(η6-C6Me6][PF6], reaction with a G4 polypropyleneimine dendrimer containing a theoretical number of 64 amino termini provided the orange polycationic FeII amido-linked metallodendrimer that was reduced to the deep-purple neutral FeI dendrimer. Further reaction with C60 gave the dendritic FeII salt of C60 − as the counter anions that were characterized by Mössbauer and EPR spectroscopies [57]. This electron-transfer reaction of a dendritic reservoir of 64 electrons is an example of the potential of metallodendrimers to serve as molecular nanobatteries [57] (Scheme 2).

Synthesis of a dendritic 19-electron complex containing 64 [(η5-C5H4 CONH-)FeI(η6-C6Me6)] termini and reaction between this dendrimer 64-dendr-[(η5-C5H4 CONH-)Fe(η6-C6Me6)]64 and C60 yielding the reduction product of 64 C60 molecules to their radical anions C60 − [68]
6 Click Chemistry as a New Tool to Assemble Redox-Active Metallodendrimers
The first example of click chemistry [58] to assemble metallodendrimers [59, 60] using the copper-catalyzed azide alkyne cycloaddition reaction (CuAAC) [61–66] was the series of reactions between dendritic cores containing 9, 27 and 81 azidomethyl termini with ethynylferrocene yielding 1,2,3-triazolylferrocene-terminated dendrimers [67, 68]. This new class of metallodendrimers showed cyclic voltammograms with a single, fully reversible ferrocenyl wave that allowed sensing oxo-anions and transition-metal cations [67, 68] (Fig. 5) as later did “reverse” click metallodendrimers constructed by reactions of a series of dendritic cores containing 9, 27, 81 and 243 propargyl termini with azidomethylferrocene [69].

With ethynylbiferrocene, the “click” CuAAC reactions of dendritic cores containing up to 729 azidomethyl termini yielded 1,2,3-triazolylbiferrocenyl-terminated metallodendrimers in which the terminal ferrocenyl units were more easily oxidized than the inner triazolyl-substituted ferrocenyl units because of the electron-withdrawing character of the triazolyl substituent [70]. The mixed-valence dendritic polycations obtained by single-electron oxidation of each biferrocenyl termini were indeed localized (Class 2 of mixed-valence compounds), as indicated by Mössbauer spectroscopy (Scheme 3).

Synthesis of the mixed-valence “click” triazolylbiferrocenium FeII/FeIII (ferrocene/ferricinium) dendrimer upon selective oxidation of the outer ferrocenyl groups of the triazolylbiferrocene dendrimer [70]
Redox recognition led to both oxo-anion sensing using the cyclic voltammetry wave of the terminal outer ferrocenyl groups and PdII sensing with that of the inner, triazolyl-substituted ferrocenyl groups [70].
In all these metallodendrimers, the absence of observable electrostatic effects lets us observe a single reversible cyclic voltammetry wave for all the chemically equivalent ferrocenyl groups in a given dendrimer, which made molecular recognition and sensing easy and simple using this technique [71–73]. This is not the case, however, with rigid hexa(ethynylferrocenyl) benzene stars, in which the differentiated proximity among the ortho, meta and para substituent of the arene maximizes these electrostatic effects inhibiting the observation of single cyclic voltammetry waves for the peripheral ferrocenyl groups [73–75].
7 Gold Nanoparticles as Templates for the Click Assembly of Redox-Active Metallocene Materials
The facile assembly of gold nanoparticles (AuNPs) with ferrocenyl-terminated alkylthiolate ligands upon reduction of HAuCl4 in the presence of thiols is an excellent method of synthesis of nano-sized assemblies with large numbers of chemically equivalent ferrocenyl groups [76, 77]. Such ferrocenyl nanosystems are also oxo-anion sensors, and this strategy has been extended to dendrons containing 3 or 9 ferrocenyl termini and a thiol group at the focal point, which led to the first nanoparticle-cored dendrimers [78, 79]. These AuNP-cored dendrimers are sensors of the ATP anion, and Pt electrodes modified with such large nano-assemblies are stable and recyclable upon washing the ATP substrate [79].
The recent study of the functionalization of nano-sized assemblies by click chemistry showed that this type of functionalization was marred by serious problems. For the construction of metallodendrimers of moderate size, the use of catalytic amounts of CuSO4 + sodium ascorbate is not sufficient because of the complexation of the copper ions to the triazolyl ligands inhibiting further catalytic activity, and the click dendrimer construction was shown to be only possible using stoichiometric amounts of this Sharpless catalyst [67, 68]. For click reactions of a nonaazido derivative, CuI catalysts with nitrogen ligand work in small amounts [80] but with dendrimers of average size, for instance with 27 azido termini, CuI catalysts with bulky nitrogen ligands do not work due to steric inhibition. The new catalyst [Cu(hexabenzyl)triaminoamine]Br, however, is efficient in catalytic (6 %) amount even for the synthesis of large dendrimers [81].
In the case of AuNPs, the situation is even worse than with dendrimers, because up to 400 % of CuSO4 + sodium ascorbate is necessary to conduct the click reaction between azido-terminated AuNPs and terminal alkynes, and the yield is modest or low due to extensive AuNP aggregation [82]. The use of [Cu(hexabenzyl)triaminoamine]Br in 10 % amount successfully led to the click functionalization of azidoalkylthiolate-AuNPs of 2.7 nm size with a variety of organic, organometallic and polymeric terminal alkynes including triferrocene dendrons [83, 84]. In the latter example, clicked AuNP-cored metallodendrimers containing approx. 150 ferrocenyl termini were indeed cleany obtained using this catalyst (Scheme 4) [83].

Functionalization of poly azido-terminated gold nanoparticles for the synthesis of gold nanoparticle-cored ferrocene-terminated dendrimers [83]
8 Conclusion: Dendritic Redox Reagents and Click Chemistry
Dendritic assemblies provide unique well-defined topologies to hold large numbers of various redox groups, ferrocene being an excellent prototype in a strategy that compares to that involving polymers that contain ferrocenes [85–88] and other iron-sandwich complexes [88]. Giant metallocene dendrimers were recently made accessible and cationic cobaltocene and CpFe(η6-arene) units have also been linked to such dendrimers. Click chemistry is an especially practical means of branching such transition-metal sandwich using appropriate CuI catalyst that take into account both the ligand protection of the metal center, solubility problems and the need to avoid steric inhibition in nano-systems. Finally, the supramolecular aspect of dendrimers is a key aspect that will allow the control of interaction with redox-robust species in green catalysts [89–91] and future devices [92].
References
F.A. Cotton, G. Wilkinson, C.A. Murillo, M. Bochman, Advanced Inorganic Chemistry, 6th edn. (Wiley Interscience, New York, 1999)
A.S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon, W. Van Schalkwijk, Nat. Mater. 4, 366–377 (2005)
I. Bertini, H.B. Gray, S.J. Lippard, J.S. Valentine, Bioinorganic Chemistry (University Science Books, Mill Valley, 1994)
D. Astruc. Electron Transfer and Radical Processes in Transition Metal Chemistry (Wiley–VCH, New York, 1995). See also the detailed foreword by H. Taube therein
D. Astruc, Nat. Chem. 4, 255–267 (2012)
K.M. Kadish, K.M. Smith, R. Guilard (eds.), Handbook of Porphyrin Science, Vol 1–25 (World Scientific Publishing, Singapore, 2010–2012)
T.P. Fehlner, J.-F. Halet, J.-Y. Saillard, Molecular Clusters (Cambridge University Press, Cambridge, 2007)
D. Astruc, Organometallic Chemistry and Catalysis Chap. 11 (Springer, Heidelberg, 2007)
Electron Transfer in Chemistry, Vol. 1–4, ed. by V. Balzani. (Wiley–VCH, Weinheim, 2001)
N.G. Connelly, W.E. Geiger, Adv. Organomet. Chem. 23, 1–93 (1984)
W.E. Geiger, N.G. Connelly, Adv. Organomet. Chem. 24, 87–130 (1985)
W.E. Geiger, J. Organomet. Chem. Libr. 23, 142–172 (1990)
N.G. Connelly, W.E. Geiger, Chem. Rev. 96, 877–910 (1996)
W.E. Geiger, Organometallics 26, 5738–5765 (2007)
P.D. Beer, P.A. Gale, Angew. Chem. Int. Ed. 40, 486–416 (2001)
O. Reynes, J.-C. Moutet, J. Pecaut, G. Royal, E. Saint-Aman, New J. Chem. 26, 9–12 (2002)
M.P.G. Armada, J. Losada, M. Zamora, B. Alonso, I. Cuadrado, C.M. Casado, Bioelectrochem. 69, 65–73 (2006)
F.J. Martinez, B. Gonzales, B. Alonso, J. Losada, M.P. Garcia-Armada, C. Casado, J. Inorg. Chem. Polym. Mater. 18, 51–58 (2008)
D. Astruc, M.-C. Daniel, J. Ruiz, Chem. Commun. 2637–2649 (2004)
R.A. Sheldon, J.K. Kochi, Metal-Catalyzed Oxidation of Organic Compounds (Academic Press, New York, 1981)
J.-M. Savéant, Acc. Chem. Res. 13, 323–329 (1980)
E. Steckhan, Angew. Chem. Int. Ed. Engl. 25, 683 (1986)
A.E. Shilov, Russ. Chem. Rev. 43, 378–398 (1974)
G. Wilkinson, M. Rosenblum, M.C. Whittig, R.B. Woodward, J. Am. Chem. Soc. 74, 2125–2126 (1952)
G. Wilkinson, P.L. Pauson, F.A. Cotton, J. Am. Chem. Soc. 76, 1970–1974 (1954)
R.B. King, Organometallic Syntheses, vol 1 (Academic Press, New York, 1965)
U. Kölle, F. Khouzami, Angew. Chem. Int. Ed. Engl. 19, 640–641 (1980)
J. Ruiz, D. Astruc, C.R. Acad, Sci. Ser. IIc Chim. 1, 21–27 (1998)
I. Noviandry, K.N. Brown, D.S. Fleming, P.T. Gulyas, P.A. Lay, A.F. Masters, L.J. Phillips, J. Phys. Chem. B. 103, 6713–6722 (1999)
J.-R. Hamon, D. Astruc, P. Michaud, J. Am. Chem. Soc. 103, 758–766 (1981)
M. Lacoste, F. Varret, L. Toupet, D. Astruc, J. Am. Chem. Soc. 109, 6504–6506 (1987)
M.H. Desbois, D. Astruc, J. Guillin, F. Varret, J. Am. Chem. Soc. 111, 5800–5809 (1989)
C. Moinet, E. Roman, D. Astruc, J. Electroanal. Chem. 121, 241–253 (1981)
S. Rigaut, M.-H. Delville, J. Losada, D. Astruc, Inorg. Chim. Acta. 334, 225–242 (2002)
J. Ruiz, M. Lacoste, D. Astruc, J. Am. Chem. Soc. 112, 5471–5483 (1990)
D.S. Brown, M.-H. Delville, R. Boese, K.P.C. Vollhardt, D. Astruc, Angew. Chem. Int. Ed. Engl. 33, 661–663 (1994)
E. Alonso, D. Astruc, J. Am. Chem. Soc. 122, 3222–3223 (2000)
J.C. Green, M.R. Kelly, M.C. Payne, E.A. Seddon, D. Astruc, J.-R. Hamon, P. Michaud, Organometallics. 2, 211–218 (1983)
A. Madonik, D. Astruc, J. Am. Chem. Soc. 106, 2437–2439 (1984)
A.N. Nesmeyanov, Adv. Organomet. Chem. 10, 1 (1972)
R.G. Sutherland, S.C. Chen, W.J. Pannekoek, C.C. Lee, Ann. N. Y. Acad. Sci. 295, 192–200 (1977)
J. Ruiz, F. Ogliaro, J.-Y. Saillard, J.-F. Halet, F. Varret, D. Astruc, J. Am. Chem. Soc. 120, 11693–11705 (1998)
G.R. Newkome, C.N. Moorefield, F. Vögtle, Dendrimers and Dendrons, Syntheses, Concepts, Applications (Wiley–VCH, Weinheim, 2001)
G.R. Newkome, C.N. Moorefield, Chem. Rev. 99, 1689–1746 (1999)
G.R. Newkome, C. Shreiner, Chem. Rev. 110, 6338–6442 (2010)
S.H. Hwang, C.D. Shreiner, C.N. Moorefield, G.R. Newkome, New J. Chem. 31, 1192–1217 (2007)
C.M. Casado, I. Cuadrado, M. Moran, B. Alonso, B. Garcia, B. Gonzales, J. Losada, Coord. Chem. Rev. 185–6, 53–79 (1999)
J. Ruiz, G. Lafuente, S. Marcen, C. Ornelas, S. Lazare, E. Cloutet, J.-C. Blais, D. Astruc, J. Am. Chem. Soc. 125, 7250–7257 (2003)
C. Ornelas, J. Ruiz, C. Belin, D. Astruc, J. Am. Chem. Soc. 131, 590–601 (2009)
C. Valério, J.-L. Fillaut, J. Ruiz, J. Guittard, J.-C. Blais, D. Astruc, J. Am. Chem. Soc. 119, 2588–2589 (1997)
C. Ornelas, J. Ruiz, D. Astruc, Organometallics. 28, 4431–4437 (2009)
D. Astruc, E. Boisselier, C. Ornelas, Chem. Rev. 110, 1857–1959 (2010)
G.R. Newkome, Z. Yao, G.R. Baker, V.K. Gupta, J. Org. Chem. 50, 1857–1859 (1985)
C. Ornelas, D. Méry, E. Cloutet, J. Ruiz, D. Astruc, J. Am. Chem. Soc. 130, 1495–1506 (2008)
T. Kusamoto, J. Ruiz, D. Astruc, New J. Chem. 33, 2204–2209 (2009)
C. Ornelas, J. Ruiz, D. Astruc, Organometallics. 28, 2716–2723 (2009)
R. Djeda, C. Ornelas, J. Ruiz, D. Astruc, Inorg. Chem. 49, 6085–6101 (2010)
H.C. Kolb, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 40, 2004–2021 (2001)
D. Astruc, F. Chardac, Chem. Rev. 101, 2991–3031 (2001)
G. Franc, A. Kakkar, Chem. Commun. 5267–5276 (2008)
V.V. Rostovtsev, L.G. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed. 41, 2596–2599 (2002)
M. Meldal, C.W. Tornøe, C. Christensen, J. Org. Chem. 67, 3057–3064 (2002)
M. Meldal, C.W. Tornøe, Chem. Rev. 108, 2952–3015 (2008)
P.L. Golas, K. Matyjaszewski, Chem. Soc. Rev. 39, 1338–1354 (2010)
J.E. Hein, V.V. Fokin, Chem. Soc. Rev. 39, 1300–1315 (2010)
L. Liang, D. Astruc, Coord. Chem. Rev. 255, 2933–2945 (2011)
C. Ornelas, J. Ruiz, E. Cloutet, S. Alves, D. Astruc, Angew. Chem. Int. Ed. 46, 872–877 (2007)
C. Ornelas, L. Salmon, J. Ruiz, D. Astruc, Chem. Eur. J. 14, 50–64 (2008)
J. Camponovo, J. Ruiz, E. Cloutet, D. Astruc, Chem. Eur. J. 15, 2990–3002 (2009)
R. Djeda, A. Rapakousiou, L. Liang, N. Guidolin, J. Ruiz, D. Astruc, Angew. Chem. Int. Ed. 49, 8152–8156 (2010)
J.B. Flanagan, A. Marcel, A.J. Bard, F.C. Anson, J. Am. Chem. Soc. 100, 4258–4253 (1978)
D.E. Richardson, H. Taube, Coord. Chem. Rev. 60, 107–129 (1984)
F. Barrière, W.E. Geiger, Acc. Chem. Res. 43, 1030–1039 (2010)
A.K. Diallo, J.-C. Daran, F. Varret, J. Ruiz, D. Astruc, Angew. Chem. Int. Ed. 48, 3141–3145 (2009)
A.K. Diallo, C. Absalon, J. Ruiz, D. Astruc, J. Am. Chem. Soc. 133, 629–641 (2011)
A. Labande, D. Astruc, Chem. Commun. 1007–1008 (2000)
A. Labande, J. Ruiz, D. Astruc, J. Am. Chem. Soc. 124, 1782–1789 (2002)
M.-C. Daniel, J. Ruiz, S. Nlate, J. Palumbo, J.-C. Blais, D. Astruc, Chem. Commun. 2000–2001 (2001)
M.-C. Daniel, J. Ruiz, S. Nlate, J.-C. Blais, D. Astruc, J. Am. Chem. Soc. 125, 2617–2628 (2003)
N. Candelon, D. Lastécouères, A.K. Diallo, J. Ruiz, D. Astruc, J.-M. Vincent, Chem. Commun. 741–743 (2008)
L. Liang, J. Ruiz, D. Astruc, Adv. Syn. Catal. 353, 3434–3450 (2011)
A. François, A. Laroche, N. Pinaud, L. Salmon, J. Ruiz, J. Robert, D. Astruc, ChemMedChem. 6, 2003–2008 (2011)
P. Zhao, M. Grillaud, L. Salmon, J. Ruiz, D. Astruc, Adv. Syn. Catal. 354, 1001–1011 (2012)
D. Astruc, L. Liang, A. Rapakousiou, J. Ruiz, Acc. Chem. Res. 45, 630–640 (2012)
Frontiers in Transition-Metal Containing Polymers, ed. by A.S. Abd-El-Aziz, I. Manners (Wiley, New York, 2007)
I. Manners, Science. 294, 1664–1666 (2001)
D. Herbert, U.F. Mayer, I. Manners, Angew. Chem. Int. Ed. 46, 5060–5081 (2007)
A.S. Abd-El-Aziz, S. Bernardin, Coord. Chem. Rev. 203, 219–267 (2000)
B. Helms, J.M.J. Fréchet, Adv. Syn. Catal. 348, 1125–1148 (2006)
D. Astruc, K. Heuze, S. Gatard, D. Méry, S. Nlate, L. Plault, Adv. Syn. Catal. 347, 329–338 (2005)
C. Ornelas, J. Ruiz, L. Salmon, D. Astruc, Adv. Syn. Catal. 350, 837–845 (2008)
Y. Ochi, M. Suzuki, T. Imaoka, H. Nishihara, Y. Einaga, K. Yamamoto, J. Am. Chem. Soc. 132, 5061–5069 (2010)
Acknowledgement
Invaluable contributions and discussions with Drs Azzedine Bousseksou, Lionel Salmon and Jean-Claude Daran (LCC, Toulouse), Prof. Jean-Yves Saillard, Drs Samia Kahlal and Christiane Perrin (Rennes), and Prof François Varret (Versailles), concerning the subject of this mini-review during the last decade, and financial support from the University Bordeaux 1, the Centre National de la Recherche Scientifique (CNRS), the Institut Universitaire de France (IUF), the Agence Nationale pour la Recherche (ANR, PhD grants to LL and AD), the China Scholarship Council (CSC) from the People’s Republic of China (PhD grant to PZ), the Fundaçao para a Ciencia e a Technologia, Portugal (PhD grant to CO), and the Ministère de l’Enseignement Supérieur de la Côte d’Ivoire (PhD grant to RD) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
This mini-review article is dedicated to our distinguished friend Professor Hiroshi Nishihara, a gentleman and superb scientist and teacher.
Rights and permissions
About this article
Cite this article
Astruc, D., Zhao, P., Liang, L. et al. Dendritic Molecular Nanobatteries and the Contribution of Click Chemistry. J Inorg Organomet Polym 23, 41–49 (2013). https://doi.org/10.1007/s10904-012-9720-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-012-9720-x