
Abstract
Supramolecular multinuclear systems with well-defined structures and architectures capable of responding to external stimuli such as photons or electrons have become increasingly interesting and are fundamental objectives for the design of future functional devices at the nanoscale. Metal-containing macromolecules as coordination dendrimers, polymers, and cages have become a field of huge and wide interest because of their possible application in a number of fields ranging from solar energy conversion and optoelectronics to bioimaging and drug delivery. The presence of transition metal ions in a supramolecular structure would offer an attractive platform for combining the chemical, electronic, and optical properties of metal complexes with those of the organic materials. In recent decades, many efforts have been devoted to the development of efficient and effective synthetic protocols for the preparation of these multi-nuclear structures with specific functions.
In this chapter we would like to resume the principal research output of the field, essentially related to the facile tuning of photophysical properties and photo-activated functions of these versatile materials. To this goal we have tried to underlie the relationship between chemical and electronic structures of these species and their photophysical properties. Because of their intriguing properties the literature is very extended, and then it is clear that is not possible to prepare a review which can be comprehensive on photoactive supramolecular multimetallic compounds, so this chapter must be limited in its scope and will deal with a few examples of coordination dendrimers, polymers, and cages.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Coordination dendrimers
- Coordination polymers
- Coordination cages
- Supramolecular chemistry
- Photophysics
- Metal complexes
- Solar energy conversion
- Sensors
- Drug-delivery
- Optoelectronics
1 Dendrimers
Chemical synthesis has long been considered a realm of organic chemistry. In the last 30 years, however, considerable progress has been made in the field of the synthesis of metal complexes, and it can now be said that, as far as the preparation of new artificial compounds is concerned, the chemistry of coordination of metals competes in imagination and realization with organic chemistry. This also applies to polymeric systems and dendrimers in particular. This class of macromolecules with a well-defined tree structure, developed for the first time by organic chemists [1], has greatly expanded its aesthetic attractiveness and its multitude of potential applications in the hands of coordination chemists [2].
Among many, self-assembly, which is based on the cooperativity of several noncovalent interactions including electrostatic, hydrogen-bonding, metal-ligand coordination interactions (bonding energy approx. 15–50 kcal mol−1), etc. has proven to be an effective approach for the preparation of multimetallic systems.
So, the metal-ligand coordination interaction is particularly attractive because the strength of the bond is relatively high and is selectively directional.
On the one hand, in fact, it provides a synthetic alternative to the carbon-carbon or carbon-heteroatom bond, on the other hand it allows to give the new structures new properties (related to the nature of the metal and the bond of coordination formed with the binders) by increasing the area of application of multichromophoric systems such as catalysts, supramolecular redox sensors, and antennas for the collection of light energy.
Moreover, the dynamic nature of the metal-ligand coordination bonds (bonding energy approx. 15–50 kcal mol−1) confers the ability to modulate the properties of these multi-metallic systems.
From the pioneering work of Newkome [3, 4] and Balzani, Campagna, Denti, and Serroni [5,6,7] in the early 1990s, the building of supramolecular metallodendrimers by choosing the appropriate ligands and metal ions has evolved to become one of the most interesting fields in the field of coordination polymer chemistry.
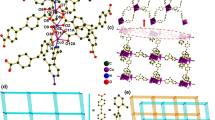
The term dendrimer (from the Greek dendron = tree) indicates a particular molecular structure that branches in three dimensions starting from a central core recalling precisely the structure of a tree. In a system of this type it is possible to distinguish, in addition to the central core, the different repetitive units that define the different generations of the dendrimer (see Fig. 24.1).

Artwork (a) and schematic (b) representation of a dendrimer structure
The growing interest in this type of system is unequivocally demonstrated by the trend in the number of publications on this sector that have appeared in the literature. At the beginning the interest was essentially focused on the synthesis of dendrimers with new skeletons and higher and higher generations; with the passing of time the research has been more and more directed to the targeted design of systems that could perform certain functions [8,9,10,11].
On the other hand, the commercial availability of some of these systems and the progressive development of analytical techniques have given further impetus to this field of research and the first examples of applications have begun to appear.
Metal complexes are characterized by a precise molecular geometry related to the coordination number characteristic of the metal ion in the given state of oxidation, to the more or less rigid structure of the ligands as well as their electronic properties.
Precious properties such as visible light absorption, luminescence and accessible reduction and oxidation potentials become an integral part of organized and controlled structures.
To design a dendrimeric system with the desired properties, attention must be paid to: (i) the nature of the chromophores and building blocks; (ii) the connections between the chromophores; (iii) the synthetic strategies that allow topological control.
Metal centers can generally be used as the core of a dendrimer by means of metal-ligand coordination bonds. Complexes [M(L)n] of metals M containing ligands L that can be easily replaced are generally used to synthesize these metallodendrimers. Among the many metals, Ru(II) and Os(II) are an excellent candidate due to their remarkable ability to form, with the proper chelators, a stable coordination.
From a structural point of view, most of the dendrimers containing metals can be classified into the following four categories:
-
1.
Dendrimers constructed around a metal complex acting as a core: these compounds may be considered metal complexes of dendritic substituting carrier binders. The most commonly used metal complexes as cores are: porphyrin complexes, polypyridine complexes and ferrocene type compounds.
-
2.
Dendrimers containing metal complexes placed as peripheral units: these dendrimers belong to the class of dendrimers functionalized on the surface [12,13,14].
-
3.
Dendrimers containing metal complexes in the branches: in these compounds, metal complexes play the role of connectors along the branches of the dendritic structure, or they can be attached to specific sites [15].
-
4.
Dendrimers in which metals occupy all branching sites: these compounds are based on the ability of ligands to coordinate two or more metal centers. In addition to metal ions and bridge ligands, they contain terminal ligands [16,17,18,19].
-
5.
There are also dendrimeric species that combine the characteristics of more than one category.
1.1 Synthetic Strategies
Dendrimers are usually prepared by divergent or convergent approaches (Figs. 24.2 and 24.3): the divergent approach involves a divergent iterative coupling and successive activation protocol from the core; the convergent approach is based on the pre-synthesis of dendrimer branches (dendrons) and the connection of the various branches to the core.
1.1.1 Divergent Synthesis
In this type of approach the dendrimer is built starting from the core for successive addition of the different repetitive units. In particular, the polyfunctional core reacts with appropriate building blocks to give the first generation dendrimer. If the peripheral units still contain reactive sites, or if reactions are possible to introduce them, the first generation dendrimer may react further and the process may be iterated to obtain subsequent generations. A representation of this type of approach is shown in Fig. 24.2.

Schematic representation of a divergent methodology
A significant feature of all divergent approaches is the rapid increase in the number of reactive sites on the periphery of the dendrimer. This can lead to potential problems in the growth process. Any incomplete reaction of the end assemblies can cause imperfections and failures in the subsequent steps. In addition, to prevent side reactions and to force the reaction to be complete, large excesses of reagents are generally used and this obviously complicates the purification processes .
1.1.2 Convergent Synthesis
This synthetic approach is shown in Fig. 24.3.

Schematic representation of a convergent methodology
In this type of approach, the dendritic structure is built from the periphery and, step by step, the synthesis is designed to use a limited number of reactive sites, thus building the ramifications that are then made to react with the core .
In order to better clarify the differences between the two types of approach and to highlight more clearly their respective potentials, some examples from literature are reported and discussed below.
1.1.3 Comparison of Divergent and Convergent Approaches
The first example of iterative synthesis was reported by Vögtle in 1978, although the main aim of the work was the synthesis of molecules that could act as receptors [20].
This kind of synthesis is based on the cyanoethylation of amines with acrylonitrile in a Michael addition reaction, followed by the chemical reduction of cyanides in order to restore amino functions in the periphery. The chemical reduction of nitriles has as a secondary reaction the breaking of the side chains. Vögtle himself then developed a simpler method of reduction [21]. In 1993 two research groups, one German and the other Dutch, published, practically at the same time, an improvement in the synthesis of polytrimethylenimines functionalised at the periphery with nitriles or amines. This procedure is based on the hydrogenation of nitriles in amines. This improvement has allowed the synthesis of similar high generation systems [22].
In the field of coordination chemistry, the divergent approach has been successfully used in the synthesis of dendrimers containing up to twenty-two metal centres [23].
The components used and their graphic representations are shown in Fig. 24.4.

Ligands used in the synthesis of selected metal dendrimers
In order to successfully conduct a divergent synthesis it would be desirable to have bifunctional building-blocks, in the chemistry of coordination a versatile building block should contain labile binders that can be properly replaced by appropriate chelating sites and coordination sites still free or protected to coordinate new metal centers.
This strategy derives from the concept of “complexes as ligand/complexes as metals” (“cal/cam”) origins of the synthetic strategy [24]. It is, precisely, an example of modular synthetic chemistry, a general approach that is now widely adopted in chemical synthesis and is based on the very concept of supramolecular chemistry. To obtain large supramolecular structures it is useful to prepare simpler (multi)component structures (modules) and insert them into structurally organized systems by exploiting some specific and absolutely complementary reaction sites.
Mononuclear metal complexes are typically synthesized by reacting, according to the scheme (M + nL ➔ MLn), suitable metal ion precursors (M) and free ligands (L). The “cal/cam” strategy is based on the use of complexes instead of simple metal ions and free ligands in both cases [24, 25].
The “M” represents mono- or oligonuclear complexes that have easily replaceable binders, so that species with unsaturated coordination sites can be generated under the reaction conditions employed (“complex metals”), and L is essentially a mono or oligonuclear complex with free chelators (“complex ligand”).
Unfortunately, a complex of this type cannot be isolated because it is a self-reactive species in the conditions of preparation (the chlorides of one unit would be replaced by the chelating sites on another), to overcome this type of problem the authors thought to protect one of the two reactive functions.
This was achieved by methylating one of the two chelating sites of the 2,3-dpp bridging ligand. This reaction transforms the bridge ligand into a terminal ligand (2,3-Medpp+, see Fig. 24.4).
The new ligand thus prepared is stable in the conditions of preparation of its complexes and it was possible to synthesize the building block shown in Fig. 24.5.

Schematic representation (a) and structure formula of [Cl2Ru(2,3-Medpp)2]2+ (b) building block
This building block proved to be particularly versatile because it was possible to first exploit the reactivity due to the presence of chlorides by making it react with appropriate bonding substrates (in conditions where the protection of the chelating site is demonstrated to be stable), were thus obtained systems “closed” containing in the peripheral positions of coordination sites “blocked” by the presence of methyl. The reactivity of these latter sites was then restored through a transalkylation reaction with DABCO that provided systems with coordination sites in the periphery again available for the reaction with appropriate metal centers. The divergent synthesis strategy developed using this interesting building block is shown in Fig. 24.6.

Schematic representation of dendrimer synthesis by divergent and convergent methodologies
It is interesting to note that any “open” complex, i.e., containing free chelating sites, as highlighted in Fig. 24.5, can be used as a core in appropriate converging approaches. For example, the mononuclear core [Ru(2,3-dpp)3](PF6)2 and the analogous Os complex – [Os(2,3-dpp)3](PF6)2 – have been successfully used in the convergent synthesis of decanuclear systems (see Fig. 24.7).

General representation of decanuclear metal based dendrimer synthesis by a convergent approach
As shown in the diagram, within these supramolecular structures, metals can occupy three different topological sites:
-
1.
Central: Occupied by the Mc metal coming from the complex used as core.
-
2.
Intermediate: Occupied by Mi metals, which occupy the central position of the trinuclear complex used as a branch.
-
3.
Peripheral: Occupied by the Mp metals that occupy the peripheral positions in the trinuclear complex.
It is therefore clear that there is a synthetic control on the topology of the system, i.e., the appropriate choice of the starting building blocks translates into the topology of the decanuclear system obtained.
The first example of a convergent approach is to be found in Fréchet work [26].
The key steps in the synthesis of the ramifications consist in the reaction of benzyl bromides with 3,5-dihydroxybenzyl alcohol. This allows the next generation of benzyl alcohol to be obtained.
On the benzyl alcohol thus obtained, it is possible to carry out a further bromination step in order to restore the bromide function and thus to iterate the growth process.
The ramifications thus obtained are reacted with a core suitably functionalized.
1.2 Photochemical and Photophysical Properties
Most of the coordination dendrimers have photophysical properties, which can be discussed within the approximation of the localized orbital molecular and which are substantially controlled by the characteristics of the metal-based units and the topology of the system [27].
Many of the examples reported in the literature are based on metal dendrimers characterized by photochemical and photophysical properties associated with luminescent and long-living excited states at adequate energies.
The energy gradient is a fundamental parameter in order to obtain e.g. energy and electrons transfer processes between the units of the same dendrimer or between the dendrimer and external species.
As already said, most of these supramolecular arrays are made by polypyridine complexes of Ru(II) [28], Os(II), and Re(I) and/or cyclometalated complexes of Rh(III) and Ir(III) [29, 30]. The use of these photo- and redox-active subunits, organized in dendrimeric structures, lies in the possibility, based on the properties of the individual building-blocks, to organize in terms of space, time, and energy the excited states in order to optimize the photophysical processes of the dendrimeric structure.
All the dendrimers discussed in this work, with some exceptions, are based on these photoactive subunits.
In general, in these compounds, the excited states involved in photochemical and photophysical processes are, by nature, the charge transfer from metal to ligand (MLCT), and or ligand to metal charge transfer (LMCT) [29, 30]. The energy of these states strictly depends on the oxidation potential of the metal and the reduction potential of the ligand [27].
As far as the structure is concerned, most of the photochemical and photophysical studies concern dendrimers of the type shown in Fig. 24.6. Only a few examples of type a and none of types b and c have been studied from that perspective. For dendrimers that contain metal-based units only in the core, an interesting issue is whether the excited properties of such units are affected by the surrounding branches. For metal-based dendrimers as branching centers, the most interesting challenges are the mutual perturbation between the chromophores and the appearance of photo-induced electron and/or energy transfer processes (intradendrimers). In most cases, these processes are highly efficient between units that share a bridging ligand, while they can be much less efficient between metal-based units that are very far apart [27]. In addition, due to the presence of many metal-based chromophores, each characterized by visible MLCT absorption bands, d-type dendrimers have significant absorption. This property, together with the control of energy migration patterns, justifies why these compounds are often considered artificial antennas [31].
1.2.1 Properties of Selected Examples
A recent review by Campagna et al. [32] resume the main photophysical properties of this kind of dendrimers that are essentially made for photoinduced energy migration across the arrays.
The rationalization of the photophysical properties of d-type dendrimers, see above, and in particular of the complexes published by Balzani, Campagna et al. needed some clarification.
Although these species can be considered supramolecular systems, in the sense that the intrinsic properties of each metal subunit are maintained, the presence of bridge ligands, see Fig. 24.7, among the metal centers has a significant influence on the properties of the multinuclear system. The properties of the individual ligands themselves vary from single-coordination to double-coordination. Specifically, the redox potential for the first reduction process is less negative by several hundred mV in bis-coordinated systems. Therefore, the energy of the 3MLCT state (which is formally a Ru → μ-2,3-dpp CT state) decreases. This behavior means that the properties of the mononuclear species cannot be considered as a model for multimetallic systems, but rather must be considered as models only systems in which the ligand is bis-coordinated.
In the same way, since the energy of the MLCT states depends on the oxidation potential of the metal center and therefore on the ligand coordinated, the subunit {Ru(μ-2,3-dpp)3}2+ appears to have an MLCT excited state higher in energy than the corresponding one localized on {Ru(bpy)2(μ-2,3-dpp)}2+ subunit, although the accept orbital of the transition is located on the same type of ligand. This translates into the fact that in the all-ruthenium dendrimers, the excited states located on the peripheral subunits have lower energy than the intermediate or central ones. Therefore the energy absorbed by all chromophores is spread outside, see, for example, Ru{(μ-2,3-dpp)Ru(bpy)2}3}3]8+ and Fig. 24.8.

Schematization, 3MLCT energy order and energy transfer processes directions in OsRu3, Ru4, Ru10, and Ru22 multinuclear dendrimers
If an Os(II) subunit is present, the driving force for the energy transfer is normally favorable in the direction of the osmium center.
As observed for the bridging ligand, the metal-metal interaction mediated by the ligand is also not negligible, so the oxidation potential of the metal centers shifts to more positive values than the corresponding mononuclear species. The same can be described for ligand-ligand interactions mediated by the metal center.
On the basis of these premises, the properties of this type of multimodally system can be explained. For example, in the dendrimer [Os{(μ-2,3-dpp)Ru(bpy)2}3]8+ (OsRu3), the energy absorbed by the peripheral Ru(II) centers is efficiently transferred to the excited state localized on the osmium(II) core [33].
For the first, second and third generation all-ruthenium dendrimers, the schematization of which is shown in Fig. 24.8, the energy transfer processes lead to the final population of the 3MLCTs located on the peripheral centers. For this type of system, the main active mechanism for the photoinduced energy transfer process is Dexter-type electron exchange [34].
If in the second generation system, containing nine polypyridine centers of Ru(II) and one of Os(II), in principle the driving force should be favorable for a unidirectional transfer towards the center, the emission spectrum of this species is dominated by the radiative deactivation of the excited states located on the peripheral Ru(II) centers and with only a small contribution by the Os(II) center (see Fig. 24.9) [17].

Schematization, 3MLCT energy order and energy transfer processes directions in OsRu9
In this case, the energy transfer from the peripheral subunits to the central Os(II) is forbidden by the presence of high energy excited states located on the intermediate Ru(II) subunits. This is even more evident in the docosanuclear species OsRu21, where there are two shells of higher energy excited states [32].
Therefore the presence of Ru(II) subunits with energies of the excited states higher than those of the peripheral and central subunits was the reason for the EnT inefficiency from peripheral to central in the dendrimers that contain more than four Ru(II) centers even in the presence of an Os(II) subunit as a potential energy acceptor [7, 34].
In most metal dendrimers, bridge binders are mainly used to connect two metal centers. An exception is the HAT ligand (HAT = 1,4,5,8,9,9,9,12-hexaazatriphenylene), which, unlike the 2,3 dpp or 2,5 dpp, is able to connect three metal centers together [7, 35]. However, the electronic coupling that provides the HAT ligand is particularly high so the energy of the π* orbital state lowers to such an extent that it becomes the orbital acceptor(s) of the excited metal-to-ligand charge-transfer state (MLCT). But the relatively low energy of these states shifts the emission to near-infrared even for compounds that contain only Ru(II) centers.
Franck-Condon factors for nonradiative deactivation processes are therefore very efficient and the excited state lifetime becomes short. This behavior becomes even more pronounced in the case of complexes containing Os(II), because, at parity of ligands, the MLCT states based involving Os(II) are lower in energy than the analogues of Ru(II).
One approach used to overcome this disadvantage was to use a bridging ligand (see L in Fig. 24.10) [36] with different electronic properties than 2.3-dpp and HAT [7, 37].

Representation of the tris-chelating ligand L
L is a tris-chelating bridging ligand based on three bipyridine-type subunits connected together by a triazine scaffold. This type of electronic arrangement causes the chelating sites of L to be decoupled so minimizing the electronic interactions between the three chelating subunits.
As a result of this, the multicoordination of L does not reduce the energy of the π* orbital to an extent that makes luminescence inefficient; therefore a system containing 6 metal Ru(II) centers and an Os(II) core was prepared, see Ru6Os in Fig. 24.11, in which the photo-induced intercomponent EnT from the MLCT states involving the peripheral Ru-based subunits to the MLCT state involving the core, has been demonstrated to be efficient and quantitative.

Molecular structure of the heptanuclear complex Ru6Os
Campagna et al. also demonstrated that dendrimers made of Ru(II) and Os(II) polypyridine subunits spontaneously self-assemble in solution so changing their photophysical properties. In particular, aggregation-driven energy transfer from higher-energy states based on Ru(II) subunits to lower-lying levels involving Os(II) subunits occurs, most likely via an inter-dendrimer mechanism also in the decanuclear mixed species discussed before [38].
A way to increase the nuclearity of these systems, keeping efficient the processes of unidirectional energy transfer, is to introduce alternative subunits characterized by high-energy excitation states. In this manner, in fact, the energy of the internal chromophores is not significantly modified, but a gradient of energy towards the intermediate subunits is maintained and the energy is, therefore, necessarily directed towards the center. To demonstrate this, the decanuclear compound 2, [Os{(μ-2,3- dpp)Ru-[(μ-2,3-dpp)PtCl2]2}3](PF6)8 or OsRu3Pt6, in which the outer layer of the metallic subunits consists of PtCl2 units, has been reported.
In this species the energy of the lowest excitation state of (μ-2,3-dpp)PtCl2 is higher than that of [Ru(μ-2,3- dpp)3]2. In fact, the excitation of the intermediate chromophores, based on Ru, of the decanuclear compound OsRu3Pt6 leads to the population of the Os nucleus with quantitative efficiency [39]. The emission 3MLCT state involves Os metal center and the ligand bridge μ-2,3-dpp, with a maximum emission at 875 nm [39].
It should also be noted that the terminal chloride ligands offer a way to further function at the periphery, or perhaps to anchor this species on a surface.
Another approach to achieve unidirectional energy transfer from periphery to the core, lie with the introduction of more chromophores on the peripheral ligands. Replacing the 2,2′ bipyridine with an organic chromophore substituted bipyridine, allows the introduction of a large number of chromophores (for example, pyrenyl subunits) at the periphery of the metal containing dendrimers. In this case, for example, hybrid dendrimeric systems have been synthesized, with six pyrenyl chromophores at the periphery [40].
1.3 Dendrimers Built Around a Metal Complex as a Core
The first photoactive dendrimer in which purely organic branches have been attached in a convergent way to a metal core appeared in 1994 [41]. The photophysical properties of this compound are very similar to those of its [Ru(bpy)2(phen)]2+ core. The perturbation induced by the dendritic branches cannot be attributed to the structure, but only to the electron donor character of the benzyloxy groups. In fact, the energy transfer from the benzyloxy groups to the central nucleus could not be investigated into detail because the absorption bands of the organic chromophoric branches are hidden by the most intense ones of the polypyridine ligands of the core.
Larger organic branches of benzyloxic type were grafted onto a nucleus of porphyrin to obtain a family of photoactive dendrimers [42]. The compound shown in Fig. 24.12 is a high generation member of this family. Photophysical investigations have shown that, in the higher generation dendrimer, the long-range photoinduced electron transfer through the dendrimer structure takes place between the porphyrin core and the methyl viologen molecules that interact electrostatically with ancillary units of the structures.

Representation of a large organic dendrimer made of branches of benzyloxic type were grafted onto a nucleus of porphyrin [42]
In general, the presence of organic ramifications does not directly influence the energy of the excited states of the Ru(II) core but the dendritic scaffold offered a strong barrier to the extinction of luminescence by molecular oxygen (O2) or other species able to react with the 3MLCT excited state (methyl viologen dication, tetrathiafulvalene…) [43].
Other studies report dendrimeric systems of this type in which the peripheral organic part is able to provide photo-induced energy transfer processes to the core. For this purpose, e.g. naphthalene was placed at the periphery [44], and the photophysical properties were characterized by the transfer of the energy absorbed by the peripheral organic chromophore to the excited state of the metal core, leading to an antenna for collecting light.
In fact, in this system, the UV light absorbed by the naphthalene subunits is efficiently transferred to the core that emits phosphorescence in the visible.
This behavior represents an important step in the sensitization of lanthanide metal ions.
Lanthanide ions have very interesting photophysical properties, but often have weak absorption bands. Thus, a dendrimer that can provide a protective shell to isolate a cation and at the same time increase the emission thanks to the transfer from the periphery to the lanthanide ion to the nucleus is of great interest. Self-assembled lanthanide-based dendrimers were prepared to demonstrate this hypothesis [45]. The authors showed that increased emissions of lanthanides and cations associated with them with the shell of the dendritic nucleus shell were observed, and an antenna effect from the periphery of the core was demonstrated to promote this process [46].
Two reviews have collected examples of dendrimers for nonlinear optical processes [47, 48]. In several cases, the whole structure possesses two Photon Absorption Properties (TPA). Moreover, alkyl ruthenium dendrimers (in which the ruthenium subunits act as connection between branches) were shown to possess third – order nonlinear optical properties [49], TPA properties [50], and even three - photon absorption properties [51, 52].
1.3.1 Dendrimer Made of Porphyrins
Dendrimers with photoactive cores were synthesized using porphyrins that demonstrated valuable properties, including their use for collecting light based on an antenna effect. Attempts to synthesize dendrimers based on these subunits posed major challenges from a synthetic point of view. Between the late 1990s and 2000, Aida and colleagues provided some important examples of the potential of porphyrin-based dendrimers for applications in a wide range of fields. For example, the construction of supramolecular assemblies with flexible multiporphyrin dendrimers metallized with Zn and bipyridyl host molecules with various chain length spacers were reported [53]. In particular, their results have shown that this type of dendrimers is able to accommodate appropriate guests capable of increasing the quantum yield of the self-assembled system. More complex self-assembled structures were obtained by the construction of segregated structures.
Arrays formed by the same dendrimers and bipyridine guests with C60-fullerene units. The photo-excitation of zinc porphyrin donors led to a transfer of electrons (PET) to fullerene receptors to generate a charge separation, which depended on the generation and number of C60 units. Self-assembled systems, including a maximum of porphyrin and fullerene units provided in the best way for the separation of charges, in this case, for a remarkable dendritic effect.
Among the different examples of photoactive dendrimeric systems, not based on polypyridine subunits of transition metals, giant multiporphyrin arrays represent equally interesting cases for research [54, 55]. The synthetic strategy that can be used is similar to that described above, and these dendritic arrays can be prepared in a convergent or divergent manner [56]. The design consists of a porphyrin connected to the multi-porphyrinic branches metalated with Zn. This provides an efficient platform for energy migration between subunits with high-energy excited states (donors) prior to transfer to the acceptor with an efficiency that decreases slightly as the number of metal-porphyrins. The efficiency of energy transfer has proven to be dramatically shape-dependent. The electron transfer seems to be more efficient with a geometry suitable for donors around the acceptor. The central part must be able to trap the energy and a highly congested architecture has proved more useful for this process [57, 58].
Recently, fibrous supramolecular coordination polymers were formed from light-harvesting multiporphyrin dendrimers and multipyridyl porphyrins through axial coordination interactions of pyridyl groups. The coordination polymers reduced intramolecular energy transfer processes within the light-harvesting dendrimer, and an intermolecular energy transfer became predominant [59].
1.4 Chemistry on the Complex
In addition to the dendrimers obtained through assembly via coordination of metal ions (“cal/cam” summary strategy), several dendrimeric species were obtained using an alternative approach [60].
The chemical transformation of ruthenium complexes after coordination has been used since their inception in the 1970s. However, there have been several alternative synonyms in the literature, for example, post-coordination. De facto the chemistry on the complex aims to customize the photophysical and redox-chemical properties of the metal subunits. It also allows the conjugation of Ru complexes with other functional molecular fragments, which is very interesting for specific recognition processes (targeting functions in life science applications) or to control the processes of energy or electron transfer induced by light or transfer in more complex systems.
This method is based on preparing polypyridine subunits of mono- or multimetallic Ru(II) bearing on the polypyridine ligands reactive sites and directly coupling these modules by means of high-performance organic reactions on the coordinated ligands. The bridge ligand that allows to assemble the different metal centers, in this way, is formed during the reaction.
Constable and collaborators have prepared a large number of Ru(II) dendrimers using this synthetic alternative, exploiting both convergent and divergent approaches.
In most cases, they used ligands such as of the terpyridine type (terpy) as chelating sites.
One of the largest systems prepared by this team is an octadecanuclear complex [61,62,63]. The synthesis was the following obtained by reacting to six equivalent trinuclear compounds, ending with a hydroxylated group (treatment with base generates the reactive intermediate), with an equivalent of hexakis(bromomethyl)benzene to generate the octadecanuclear composed of a converging path [24]. Similar systems have been studied by Juris and colleagues, which used terpyridine analogues for the preparation of the first generation of a tetranuclear dendrimer mixed-metal complex [64]. In particular the synthesis of the latter compound is an example of coupling between the “cal/cam” approach and the performance strategy organic reactions on complex preforms. Starting from the alkylation of decorated bromine complexes [Ru(bpy)3]2+ with hydroxyl bpy, subsequently, a coordination reaction was carried out to generate the peripheral complexes [M(bpy)3]2+ (M = Ru, Os). Unlike previous more strongly coupled polynuclear assemblies, the photophysical properties of the subunits were mainly preserved, which aroused great interest in subsequent years. The Constable group systematically studied the synthesis of polynuclear metal-based complexes through the chemistry of alkylation [65,66,67,68]. This strategy was further applied to assemble molecular dyads, triads, etc. Although most of these sophisticated architectures contain conjugated aromatic connection models and are often assembled before the coordination, the use of the alkylation reaction is reported. For example, free or containing porphyrins have been paired with decorated bis-phenol [Ru(tpy)2]2+ to form symmetrical trinuclear systems [69].
MacDonnell and his collaborators also used the method of coupling reactive sites on different polypyridine complex as ligand to generate chirally controlled first- and second-generation metal dendrimers [70, 71]. Using chirally resolved subunits, enantiomerically pure dendrimeric species have been obtained, since the reactions occur with maintenance of the configuration in the metal centers (which are not involved in the reaction process) and the new binding agent generated by the reaction does not introduce further elements of chirality in the final species.
Recently, assemblies formed by using the strategy of linking the metal units by biphenyl units appears in literature. This strategy offers only a limited degree of conjugation between metal centers, with individual units retaining native excited states properties. So an octanuclear mixed metal compound of composition Ir7Ru having a dendrimer wedge-like structure has been prepared showing an excellent and direction antenna effect [72].
2 Coordination Polymers
Synthetic organic polymers have played a key role in the development of the economy of modern society. Although initial research was essentially focused on the field of basic thermoplastics and elastomers, towards the end of the twentieth century the focus shifted to special materials or materials with advanced and specific properties. To this end, thanks also to the progress of coordination chemistry, the focus has been on metal centers, which in addition to providing a spatial arrangement, more or less rigid, controlled, are able to give access to specific properties and functions. The incorporation of metal centers into the skeleton of polymers has led to the development of a wide range of new materials based on organometallic and coordinating compounds for potential applications in different research fields, ranging from energy storage/conversion to bioactive materials [73].
The encounter between these two worlds of chemical synthesis, has allowed the development of new materials easily tunable through the incorporation of metal centers in chains of synthetic polymers. The systems obtained, however, were in most cases either short chain or insoluble and therefore difficult to characterize.
The progress in synthesis achieved in the 1990s has made possible to prepare this type of material in relevant quantities, with a wide variety of structures, and has allowed the detailed study of their properties [74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89]. Metallopolymers can contain a variety of metal centers, from the main group metals such as Sn and Pb, to transition metals such as Fe, Ir, and Pt, to lanthanides such as Eu.
There have been several attempts at classification, but among many, the one carried out by Rehahn and Ciardelli et al. at the end of the 1990s [90, 91] distinguishes metal polymers according to their topology. According to their classification (see Fig. 24.13) three main typologies can be distinguished.

Schematic representation of Type I, II and III coordination polymers
Polymers of type I, in which metal ions / complexes are bound to the polymer in the lateral chain or in the final group by electrostatic interaction (a), coordination bond (b) or covalent bond (c). Type II polymers that have metal centers at the beginning of the main chain, where the metal centers are incorporated by covalent bonds (a) or metal-ligand coordination (b). For these two types, the electronic properties of the metal can influence the properties of the material in the most effective manner [92]. Finally, in type III polymers, metal ions are incorporated into a polymer matrix through physical interactions, the driving force for the formation of these materials is the negative free energy as a result of the chelated effect of the polymer.
Therefore, the properties of the new materials, in addition to the nature of the components can be varied by their location and by the binding type. The bonds that couple metals can be covalent, which leads to an essentially irreversible or “static” bond, or of a noncovalent type, which, as already seen for dendrimers, can allow a potentially reversible bond.
If the first synthetic approaches were aimed at the possibility of preparing soluble and high molecular weight materials in order to clarify their properties, industry research has led to the identification of the combination of metal, binder group and spacer groups such as to achieve the desired function and then prepare the required material.
Wong et al. reported about the principal properties of metallopolymers [93].The main topic of this paragraph lies with photoactive polymeric coordination materials, and in particular with Type-I and Type-II polymer.
2.1 Zn(II) Polymers (Type-II Polymers)
Among the many metal ions used to assemble coordination polymers, zinc ions have found numerous applications thanks mainly to their low cost due to high availability, low toxicity [94].
The zinc(II) ion thanks to its electronic configuration does not take part directly in the photophysics of the assembled supramolecular species, but affects those of the organic subunits binding that connects.
For example, zinc (II) complexes with Schiff-based ligands and in particular chromophore as salen-Zn(II) type complex (salen = N,N-etilenebis(salicylimine)) required to assemble Zn(II) bis(N-alkyl salicylaldiminate) have excellent fluorescent properties with exceptional photoluminescence properties, high quantum yields and good thermal stability. Moreover the synthetic strategy used to obtain these species allows to produce structural modifications useful to modulate the properties of materials [95].
A variety of systems containing Zn(II) ions with high photoluminescence quantum yields (ϕ) and electroluminescence performance (EL) have recently been synthesized. Their photophysical data are summarized in Table 24.1.
2.1.1 Zinc (II) Polymers Based on Schiff-Bases
Che et al. have first reported the preparation of polymers based on these subunits [96]. The obtained species have the characteristic spectral profile with absorption bands in the UV region of the electromagnetic spectrum assigned to transitions π➔π* centered on the binders (LC) which in this case involve the p-phenyl rings and the salicyl-aldimine ligand, only a few intense contributions are observable in the visible region within 430 nm. These species are luminescent from single 1LC states and the emission spectra extend from blue to yellow in DMF solution (e.g., blue for 1b, green for 1a and 1c, and yellow for 1h with λem at 458, 508, 501 and 562 nm, respectively). Just as the color of the emission depends on the type of binder used, the quantum luminescence yield also varies from 2% for 1e and 1h to 34% for 1f. These species can be used and are luminescent not only in solution but also when transformed into thin films (Fig. 24.14).

Structural formulae of salen-Zn(II) type polymers of the 1 and 2 series
Cao et al. in addition to studying the effect of the substitution on the nitrogen atom, have investigated the effect of the introduction on the bridge of an aryl type spacers [97]. The main effect is an increase in the emission intensity and, in addition, moving from 2a to 2c, the maximum emission gradually shifts to red. This behavior is consistent with the increased conjugation of polymers using bridge units of the fluorene, carbazole and phenyl type (emission at 530, 540, and 545 nm, respectively).
The number of examples that have appeared in the literature is enormous and some of the results are summarized in Table 24.1.
2.1.2 Zn(II) Polymers Based on Polypyridine
In 1995, Constable proposed that coordination polymers could be obtained by adding metal ions to the suitably functionalized terpyridine (terpy) [98].
In 2002, Würthner proposed the introduction of perylene bisimide-type chromophores as a bridge between two terpyridine subunits able to assemble coordination polymers using Zn(II) [99]. The introduction of redox and photoactive systems to bridge the coordinating fragments underlines their potential to be used as new functional and stable optoelectronic materials. The stability of the polymer obtained is mainly due to the fact that terpyridine ligands form stable coordination compounds with the Zn(II) ions via dπ → pπ* bonding. Therefore, geometry, stability, directionality and modularity of organic bridges allow to introduce charge transport properties with high thermal stability and flexibility in the design and structural modification, making these species important for use in optoelectronic devices (Fig. 24.15).


Structural formulae of some [Zn(tpy)2]-based metallo polymers
As previously pointed out, thanks to the d-electron shell filled, the Zn(II) ions are not directly involved in any electronic transition in this type of systems. Therefore, the electro-optical properties of metal polymers can be easily modulated according to the type of chromophore or with spacer units introduced between the chelating sites.
In 2003, Che (3a to 3i) carried out for the first time a systematic study on the exact relationship between the structure and the photophysical properties of these systems [100]. In fact, a series of terpyridine-based zinc (II) polymers have been prepared and studied using rigid structures and terpyridine base coordinators. The absorption spectra of this type of polymer are similar to each other with absorption maxima in the UV region attributed to LC transitions. The rigid spacer subunit essentially influences the photophysical properties, for example, the emission colors of 3a, 3f, 3g, 3h, and 3i, ranging from blue, to green to yellow, with emission maxima ranging from 450 (0.55) to 567 (0.55) nm and quantum yields of the order of 50% demonstrating that polymers exhibit higher quantum yields than their equivalent monomers.
Further investigation on this family of compounds led Schubert et al. to prepare similar systems with aromatic subunits with electron-donators in the bridge (for 3j to 3s) [101]. Such studies have shown that metal-polymerization leads to a shift in the wavelength of absorption due to inductive effects caused by metal ions, and, in general, a hypochromic effect. Metallopolymers with extended π-conjugated systems (i.e., 3n and 3q to 3s) present absorption bands shifted to minor energy in the range 420–450 nm. It is therefore confirmed that the displacement of the absorption bands of the metal polymers can be predicted by changing the type and extension of the π-conjugated system to bridge between the bis(terpyridine). In these particular cases, a parasite effect has been highlighted, i.e. a decrease in the quantum yield of emission accompanied by a band widening attributable to the formation of aggregates and/or excimer. Aggregation can be inhibited by introducing substituents on the bis(terpyridine) binders as observed in 3m, 3n, 3p, and 3q [102]. The energy of the emission depends directly on the extension of the conjugated system. The luminescence shifts from green to red, for example in 3k, with the increase of the effective extension of π-conjugation [103, 104].
Over the years, the number of polymers based on Zn2+ has increased, but the actual property of the polymer depends essentially on the properties of the ligand used to assemble it.
2.2 Ru(II) Polymers (Type-I Polymers)
Partly different discussion can be done for metal polymers containing Ru(II) ions.
As already discussed, the properties of the polypyridine complexes of Ru(II) can be easily modulated, therefore the use of these ions leads to the formation of metal polymers whose properties are defined both by the ligand and by the ion which is directly involved in the electronic transitions. There are two types of coordination of Ru complexes on the polymer chain. Bpy ligands are directly incorporated into the skeleton of the π-conjugated polymer or bound to the polymer as a pendant group via conjugated or nonconjugated linker. Preformed polymers in which free bipyridine subunits are present are characterized by a conjugation of the non-metallized polymer interrupted by the torsion of the inter-ring to the bpy group. However, because of the coordination of the Ru, the nonplanar bpy units are forced to a syn-planar conformation that increases the extension of the conjugation. This leads to a direct change in the photophysical properties (e.g., absorption shifted to red with higher intensity of the low-energy bands, emission bands shifted to red and lower for example) and reports the presence of the coordinated metal ion. Bipyridine ligands are directly incorporated into the skeleton of the π-conjugated polymer or bound to the polymer as a pendant group via conjugated or nonconjugated linker. Yamamoto and collaborators synthesized the first fully conjugated Ru(II)-polymers (Ru1) containing ruthenium chromophores that are directly part of the polymer chain through coordination of the bipyridine present as a repetitive unit on the polymer chain (Fig. 24.16).

Structural formulae of some Ru(II)-based type-I polymers containing bipyridine (Ru-1a) and bipyrimidine (Ru2-a) moieties
Some results are summarized in Table 24.2. The bipyridine and 5.5′-bipyrimidine (bpym) units can be inserted as coordination sites directly on the polymer chain [105]. Controlled polymerization (chain length ~10 repetitive units) is necessary to preserve good polymer solubility. The absorption spectrum of these species is characterized by the typical bands of Ru(II) polypyridine complexes. For example, Ru-1a shows an absorption band in the visible, attributed to a singlet metal to ligand charge transfer transitions (1MLCT) at 450 nm, and intense absorption bands in the ultraviolet due to π-π* transitions centered on the ligands and the polymer skeleton. Ru-1a photoluminescence is centered at 694 nm, which lies below the energy of the parent [Ru(bpy)3]2+ (emission centered at 612 nm). The energy of MLCT transitions depends on the oxidation potential of the metal and the binding agent reduction potential. So, the increased delocalization of the polymer chain makes this more easily reducible and therefore the orbital interposed by the low-energy transition involves this fragment. The absorption spectrum of the bipyrimidine-based Ru-1b polymer has an MLCT band at 435 nm and there is also a tail in the red region of the spectrum which suggests the existence of 1:2 complexes in which two metal units are coordinated to the same bipyrimidine (bpym) subunit (i.e., [(bpy)2Ru(μ-bpym)Ru(bpy)2]4+). This polymer is not photoluminescent.
Chan and his team reported on poly(benzobisoxazole) (Ru-2a and Ru-2b) and poly(benzobistiazole) (Ru-2c and Ru-2d) containing [Ru(bpy)3]2+ units, Fig. 24.17 [106]. Spectroscopy studies show that when the 2,2′-bipyridine group is incorporated into the polymer chain using the 5,5′ position, electronic delocalization is more extensive than when using the 4,4′ position. The incorporation of the ruthenium ions into these polymers dramatically increases the mobility of the charge carriers, demonstrating that Ru complexes can improve the transport of the charge through the polymeric material. Yu and colleagues have synthesized the first poly(p-phenylene vinylene)s (PPV) containing Ru(II) chromophores that are incorporated into the polymer through the bpy unit [107]. The absorption spectrum of Ru-3a exhibits the spectral characteristics typical of metallized and non-metallized polymers with absorption in the visible centered at 450 nm. The all-Ru(II) polymer Ru-3b shows an intense absorption at 550 nm, which is significantly red-shifted compared to the similar Ru-3a complex and to that exhibited by [Ru(bpy)3]2+. However, unlike the Ru-monomer polymer and organic-based polymers, these types of Ru polymers are not emissive [108]. In general, this type of polymer has been extensively investigated for applications in DSSc devices. Therefore, the interest was focused on further modulating the optical and electronic properties of these metal-organic PPV materials by incorporating Ru(II) complexes with different ligands, for example dichtonate and hydroxyquinoline (Ru-4) (Fig. 24.18).

Structural formulae of some Ru(II)-based type-I polymers containing poly(benzobisoxazole) (Ru-2a and Ru-2b) and poly(benzobistiazole) (Ru-2c and Ru-2d) subunits

Structural formulae of some Ru(II)-based type-I polymers containing poly(p-phenylene vinylene)s moieties
The replacement of dichtonate and hydroxyquinoline groups in the ancillary ligands of the Ru substantially lowers the redox potential of the Ru(II/III) with respect to similar polypyridylic complexes. This decrease in Ru(II/III) potential induces a significant red shift in the MLCT Ru → bpy-PPV MLCT transitions. The introduction of a donor-acceptor stilbene derivative (D-A) acting as a nonlinear optical chromophore as in Ru-5 has been investigated too [109]. The use of PPV polymers containing Ru as a light emitter has been reported by Chan et al. (Ru- 6), Fig. 24.19 [110]. Most of the polymers mentioned above exhibits three absorption bands which are due to the intraligand transitions centered on the bpy ligands at 290 nm, the LC transition of the conjugated main chain (410–450 nm) and the MLCT transition of the Ru complex at about 500 nm. The absorption maxima of the latter two bands are shifted to the shorter wavelengths but with greater intensity when the metal ions content is increased.

Structural formulae of some Ru(II)-based type-I polymers containing poly(p-phenylene vinylene)s chains and donor-acceptor subunits
Guillerez et al. conducted a detailed photophysical investigation of Ru-conjugated polymers based on polyphenols. Ru-7 polymers containing 3-(octythiophene) tetramers or hexamers in which organic subunits are interspersed with type- [Ru(bpy)3]2+ subunits have been reported [111]. The absorption spectrum of Ru-7b has many characteristics similar to those of Ru-8a; however, the absorption intensity observed in these species is caused by the increase in the number of thiophene rings in the repetition unit. These two polymers are weakly emissive and the Ru-7a, and the luminescence lies at energy similar to that of [Ru(bpy)3]2+. Ru-7b has an extremely weak emission centered at 770 nm. This emission is shifted to the red and significantly weaker (about 20 times) than the emission of Ru-7a.
Another case of polymers containing Ru(II) is that of nonconjugated species containing the Ruthenium based chromophore with a rigid main chain [112] (Fig. 24.20).

Structural formulae of some Ru(II)-based type-I polymers containing poly(p-phenylene vinylene)s chains of the Ru-6 to Ru-10 series
In these species, the conjugation is interrupted by metal coordination complexes; therefore, the absorption and emission characteristics are similar to those of the precursors and individual fragments of the polymer chain. Ru-9 shows an absorption pattern similar to that of the monomer (MLCT transition to 507 nm emission) [113]. Unlike Ru-3 polymers showing MLCT absorption bands centered at ~450 nm, this absorption was not observed in Ru-9 because of the orthogonal geometry between the two terpyridine binding fragments and the π-conjugated system on the perturbed main polymer chain. The absorption spectrum of Ru-7b has many characteristics similar to those of Ru-7a; however, the absorption intensity observed in these species is caused by the increase in the number of thiophene rings in the repetition unit. These two polymers are weakly emissive and the Ru-7a has a form of energy and bandwidth similar to that of [Ru(bpy)3]2+. Ru-7b has an extremely weak emission at λmax = 770 nm. This emission is shifted to red and significantly weaker (about 20 times) than the emission of Ru-7a.
Ru-8 are polymer based on tridentate 2,6-bis(benzimidazol-2-yl)pyridine ligand [112]. The polymerization brings to a new absorption band centered at 490 nm, at higher energy with respect to the case of analogous bpy-based polymer because of the less extended conjugation along the polymer chain. Moreover the absorption of benzimidazolyl-based Ru-8a species is more red-shifted with respect to benzothiazolyl based Ru-8b compounds.
Another case of polymers containing ruthenium is that of nonconjugated species containing the Ru(II)-based chromophore within a rigid main chain.
In these species the conjugation is interrupted by metallic complexes (e.g. Ru-terpyridine), amides or imides. Therefore, the absorption and emission characteristics are similar to those of the precursors and individual fragments of the polymer chain. Ru-9 shows an absorption pattern similar to that of the monomer (MLCT transition to 507 nm emission) [113]. Unlike Ru-3 polymers showing MLCT main chain absorption bands centered at ~450 nm, this absorption was not observed in Ru-9 due to the orthogonal geometry between the two terpyridine binding fragments and the π-conjugated system on the perturbed main polymer chain. The major application of this type of system is in the field of photovoltaic cells [114] The synthesis of different aromatic polyamides with 2,2′- bipyridyl moieties (Ru-10) has also been reported [115]. In this case, the luminescence is dominated by the emission from the 3MLCT states, characteristic of the [Ru(bpy)3]2+ -like subunits, in the range 680–700 nm. The nonconjugation of the polymer chain results in the fact that the emission energies are not affected by the monomer ratio.
Ru(II) complexes can also be fixed as side chain chromophores not conjugated to main chain polymers. If the main chain is a luminescent conjugated polymer, both the metal complex and the polymer can function as a light emitter and in most cases do not affect each other except by energy transfer in the excited state. Ru-11a and b belong to a series of PPV polymers with [Ru(terpy)2]2+ (Ru-11 a) or [Ru(bpy)3]2+ (Ru-11 b)pendant, Fig. 24.21 [116]. The UV-vis absorption spectra of the polymers show intense absorption bands due to both the conjugated main chain and the Ru(II) portions and resemble the typical absorption profiles exhibited by Ru(II) polyrididine complexes. Their photoluminescence spectra depend essentially on the metal complex [117].

Structural formulae of some PPV polymers in wich Ru(II) complexes are fixed as side chain chromophores and not conjugated to the main chains
2.2.1 Iridium (III) Polymers (Type-I and Type-II Polymers)
The preparation of polymers containing phosphorescent Ir(III) complexes has attracted increasing attention in relatively recent years, also because Ir(III) systems are widely used in the field of optoelectronic materials. The preparation of polymers containing phosphorescent Ir(III) complexes has therefore attracted increasing attention in relatively recent years.
In fact, the use of organic polymers for the realization of electroluminescent devices is well known, as well as the dispersion of dyes in their matrix. But the use of simply mixed systems typically suffers from phase separation, which leads to a rapid reduction in the efficiency of the devices made. An effective solution to this problem has been the introduction of Ir(III) complexes in the main and/or lateral chain of the polymer by means of a chemical bond in order to both inhibit phase separation and, from an efficiency point of view, limiting the interaction between the host chromophores, to reduce the triplet-triplet annihilation phenomena. Efficient green, red and white emission polymer-based PLEDs with main chain Ir(III) complexes have so far been successfully manufactured.
Ir(III) phosphorescent polymers, as we have seen for Ru(II) polymers, are based on two components, namely the polymer host and the Ir(III) complex host. The polymer skeleton can be conjugated or not conjugated in nature. The conjugated host polymer contains are normally based on polyfluorene such as blue emitter and excellent charge transporters, or on carbazole for the transport of the holes. Most of nonconjugated systems are based on poly(N-vinylcarbazole) (PVK) and its derivatives and have high energy blue-emissive excited state, favorable film-forming properties, durability at high temperature, and excellent hole mobility. For the phosphorescent Ir(III) complex host, the optimization of ligand structures and the relative amount of Ir contained in the polymer greatly affects the color of emission of the resulting structure. Table 24.3 collects the photophysical data of some Ir containing polymers.
2.2.2 Polymers Conjugated with Ir(III) Complexes on the Main Chain
Polymers conjugated with Ir complexes in the main chain can be divided into three types, Ir complexes anchored with ligands N∧N, C∧N or O∧O (Figs. 24.22, 24.23, 24.24, 24.25, 24.26, and 24.27). The photoluminescence properties of these Ir polymers can be varied by different approaches.

Structural formulae of some phosphorescent Polymers conjugated with Ir(III) complexes on the main chain (Ir-1 to Ir-5)


Structural formulae of some Polymers conjugated with Ir(III) complexes on the main chain (Ir-6 to Ir-9)

Structural formulae of some mixed Ir(III)-Polymers (Ir-10 and Ir-11)

Structural formulae of some mixed Ir(III)-Polymers (Ir-12 and Ir-13)


Structural formulae of some Polymers containing neutral Ir(III) complexes based on O∧O ligands (Ir-14 to Ir-17) and polymers with phenylpyridine Ir(III) complexes (Ir-18 and Ir-19)

Structural formulae of some Polymers conjugated with Ir(III) complexes in the side chain
2.2.3 Polymers Conjugated with an C∧N or N∧N or N∧N Ligand of Ir Complexes on the Main Chain (Type-II Polymer)
The behavior exhibited of these two polymers is absolutely similar. Their absorption spectra are dominated by the bands attributed to transitions essentially centered on the polymer skeleton in the UV region, while only a small contribution of the Ir(III) complexes was found in the range of 300–430 nm. Obviously the extent of the contribution in iridium absorption is a function of the relative content of Ir complexes.
Emissions are dominated by the emission of Iridium complexes (600 and 630 nm) with only a small contribution to higher energy. But this behavior depends on the percentage of Ir complexes that are introduced into the polymer. At low Ir content, it increases the high-energy emission of the polymer. Therefore, by varying the ratios between the organic part and the metal complex, it is possible to modulate the emission color. A change in the color of the Ir polymer (red) from the Ir monomer (green) has been attributed to the elongation of the conjugated ligaments of 2-phenylpyridine (ppy) during polymerization. Similar observations have been shown in Ir-3, which emits light at about 610 nm [90].
The use of poly(fluorene-carbazole) (with a max absorption at 350 nm) leads to the formation of Ir(III) polymers in which the emission is almost completely independent of the metal complex content.
For acetyl-acetonate (acac) ligand carrier complexes, the conjugation of the polymer backbone is interrupted, this results in a blue-shifted emission although the Ir complexes with acac would normally give a peak of luminescence shifted into the red. Lin et al. reported Ir-5 polymers involving benzimidazole-based Ir complexes with a different molar monomer ratio than the organic fragment [118, 119]. Again, absorption is dominated by the backbone of the polymer and benzimidazole in the 340–380 nm region. The presence of 3,6-carbazole interrupts the delocalization of electrons along the polymer and the absorption bands are shifted to shorter wavelengths. Weak MLCT absorption bands appear at 400–500 nm. The efficiency of energy transfer seems to be greater when the carbazole ratio contained in the organic part of the polymer is greater (i.e. Ir-5f to Ir-5h are more effective than Ir-5b to Ir-5d). In this case, more residual blue emissions are evident in the PL spectra from Ir-5b to Ir-5d (Fig. 24.22).
The use of copolymers (Ir-6) (Fig. 24.23) with picolinate as the ancillary ligand leads to the appearance of a new absorption peak at 420 nm.
The introduction of a O∧O ligand of neutral Ir complexes on the main chain of the polymer leads to the repetition of delocalization on the skeleton of the polymer. Therefore, in most cases the photophysical properties of these species are similar to those of the monomeric species Ir-14 [120] and Ir-15 (Fig. 24.26) [121]. The emission of this type of Ir polymers shows only peaks slightly shifted towards red compared to the emission of the corresponding organic portion. This approach offers an additional advantage in controlling the emission wavelength of chelating copolymers. Instead, the Ir-14 electroluminescence spectra show a unique triplet emission even with the low ratio of Ir to the organic portion (Fig. 24.26).
In addition to the linear polymers of the main Ir chain, the highly branched polymers Ir-18 and Ir-19 can reduce aggregation phenomena and the formation of excimer, so, if used in the solid state, the films obtained are of good quality, and consequently the emission efficiencies can be improved. Ir-18b branched hyper-branch copolymers emit green light at about 520 nm compared to the linear analogue that emits in the yellow [122, 123] (Fig. 24.26).
2.2.4 Polymers Conjugated with Ir(III) Complexes in the Side Chain (Type –I Polymers)
Polyfluorenes or fluorene-cocarbazole copolymers are often used as main polymer chains. Polymers conjugated with Ir complexes in the side chain can be manufactured by grafting in the C9 position of fluorene or in the N position of carbazole through a spacer as long alkyl chain. Up to now, red and white emission conjugated polymers have been manufactured with Ir complexes in the side chain [124, 125]. The use of isoquinoline, Ir-21 and Ir-22 (Fig. 24.27), allows to obtain red emission (595 nm) in solid systems such as PLEDs and it has been demonstrated that this emission is independent from the use of copolymers. This also indicates that the decoration of the polymer skeleton with these pendants of Ir complexes in the lateral chain has not altered the electronic properties of the polymer itself. Red emission was also obtained by using 2,4-diphenylquinoline as the chelating ligand, Ir-23. But the use of fluorinated ligand, Ir-23b and Ir23c, shifts the emission to blue. Combining the blue emission of polyfluorene backbone, and red emission complexes, it is possible to realize white PLEDs [126].
2.3 Pt (II) Containing Polymers
Like iridium, the complexes of the heavy ion Pt (II) allow a good mixing of excited states of single and triplet, high phosphorescence yields, and relatively high luminescence lifetimes. Therefore the introduction of these on polymeric chains is particularly attractive. Two main types of polymers are commonly found, one consisting of linear polymetallines in the shape of a Pt rod and the other containing cycloplatinated microorganisms.
2.3.1 Platinum(II)-Containing Metallopolyynes (Type-II Polymers)
In the field of synthetic polymers containing Pt, Pt(II) polymers with rigid bars have aroused enormous interest. A wide range of soluble Pt(II) derivatives incorporating various conjugated ring systems have been reported with different photophysical properties to make them particularly interesting, among other things, in the field of molecular optoelectronic applications. The basic structure of polyplatinynes is shown in Fig. 24.28, 24.29, and 24.30.

Schematic representation of Pt-4 species: Platinum(II)-containing metallopolyynes (Type-II polymers)

Schematic representation of Pt-5 species: Platinum(II)-containing metallopolyynes (Type-II polymers)

Schematic representation of Pt-6 species: Platinum(II)-containing metallopolyynes (Type-II polymers)
As observed for iridium, a good methodology for modulating the optical properties of these materials is to increase the delocalization on the ligands bridging the metal centers. In this case the effect is more radical, since the delocalization of electrons can be prolonged along the polymeric chain by the conjugation between the orbitals of the Pt and those of the ligands. Another way is to localize the electronic density using an unconjugated or weakly conjugated system. This type of polymer generally has high-energy triplets, in general, the functionalization on the organic skeleton and the control of D-A interactions between the bridge units allow a fine modulation of the electronic and morphological properties. Therefore, by choosing appropriate organic bridges, these metallopolymers can be luminophores in a wide wavelength range, see Table 24.4. In general, the absorption characteristics of the Pt systems are mainly assigned to the 1IL π➔π* transitions of the metal-conjugated chromophore to conjugated acetylide. The Pt-1a polymer with the phenylene spacer absorbs at 380 nm [127,128,129], and conjugation is increased by doubling the number of C units on each side of the phenylene ring in Pt-1b, leading to a red shift in absorption and emission length from 380 to 396 nm [130, 131]. As the length of the oligothiophene increases, delocalization increases and absorption transitions shift to low energy. In parallel, the emission shifts to red, Pt-2a to Pt-2c [132]. The ratio of the subunit of Pt to the organic spacer causes the energy of the triplet emission to show a red shift along the series with a rapid drop in the intensity of the triplet emission (i.e. triplet energy of Pt-2a > Pt-2b > Pt-2b > Pt-2c). A series of Pt(II) Pt-3 polyynes [133,134,135] connected by oligopyridine with reduced conjugation has been reported. Excited state transitions can be adjusted by locating the electronic density in discrete regions of the molecules. The degree of conjugation becomes significantly poor in the presence of a silyl unit (Pt-4a) [136], but this is useful for improving the luminescence properties in terms of quantum yield, compared to the more conjugated Pt polymers mentioned above. Pt-4b also reported a very high efficiency of triplet emission [137,138,139]. The work was then extended to other polyplatines containing oligo(arylene ethynylenesilylene)s and oligo(arylene ethynynylenegermylene)s with intriguing effects. A particularly interesting effect is observed through the functionalization of the nine positions of fluorene, which allows in fact to easily adjust the photophysical and electronic properties of Pt6 polymers. It should be noted that the rigid unit of planar biphenyls of fluorene guarantees a higher degree of conjugation, thus improving the luminescence effectiveness of the materials compared to the derivatives of p-phenylene [140,141,142,143,144]. These metallic materials can show a variety of colors and values, for example, in solid state.
By doubling the length of the acetylene chain from Pt-6a to Pt-6h, a red shift was observed for both absorption and phosphorescence maxima [145].
3 Coordination Cages
The interaction between bridging ligands and metal centers, if properly designed and under appropriate conditions, can generate so-called coordination cages. Essentially, metal ions and bridging ligands combine to form a pseudospheric closed structure with a central cavity, becoming a class of compounds of great interest in supramolecular chemistry in recent decades [146].
One of the first examples to appear in the literature was the M4L6 supramolecular system with a metal ion at each vertex of a tetrahedron and a bridge binder that defines each of the six edges of the structure [147, 148]; in little more than a decade Fujita’s “nanospheres” based on Pd ions have appeared [149, 150]. The structure obtained is highly symmetrical [151, 152] and therefore the very principles of chemical symmetry are able to dictate the guidelines for the implementation of self-assembly processes [153, 154].
Research in the field has evolved and attention has been paid to the useful functional properties of these cages. These are often associated with their ability to bind small molecules as hosts in the central cavities, and the development of coordination cages in which at least one of the components is photophysically active.
The particular attraction in this context is that the cage structures allow for the assembly of a large number of photoactive units surrounding a host molecule in the cavity. Such a high local concentration of chromophores in close proximity to a host that can interact with the excited states of the chromophores is something that promises exciting developments in the field of “supramolecular photochemistry”.
This section therefore examines a range of types of luminescent cages and discusses examples of emerging applications and useful functional behaviors.
As already observed for dendrimers and metal polymers, also for coordination cages, the photophysical properties depend strongly on the type of metal ion and therefore on the type of metal complexes at the top and/or on the binders that put them in communication. Therefore, the use of luminescent complexes of [Ru(II), Os(II), Re(I), Ir(III), Pt(II), Pt(II), etc.] and in general of ions with electronic configurations d6/d8 with low spin are particularly interesting. But, as already discussed, these ions, in some cases more than others, are particularly kinetically inert to provide self-assembly. Nevertheless, for example, Lusby and colleagues reported the preparation of an octahedral coordination cage in which six units of {Ir(ppy)2} at the top of the cage are connected by four 1,3,5-tricyanobenzene ligands (Fig. 24.31) [155]. This system is luminescent from the 3LC type excited state typical of subunits {Ir(ppy)2X2} (λem = 570 nm and ϕ =0.04). It has been demonstrated that the luminescence of this species can be modulated by the presence of anions inside the cavity [155].

Schematic representation of an octahedral coordination cage in which six units of {Ir(ppy)2} at the top of the cage are connected by four 1,3,5-tricyanobenzene ligands
Systems based on luminescent complexes of inert ions have been obtained for example with Ru(II) and Os(II) using particular ligand systems and the principle underlying the cal/cam strategy. In fact, a precursor based on these ions has been prepared, in which a ligand contains, in the right geometry, a free coordinating site. This “complex ligand” is then combined with a second (labile) ion, and the assembly of the free binding sites around the labile ions leads to the formation of a molecular cage [156, 158].
Systems based on subunit type [Ru(terpy)2]2+ with four ancillary pyridine units have been used, in combination with Pd(II) ions to obtain efficiently and quickly cages in which three or four of the complex units Ru(II) combine with six or eight Pd ions through the formation of Pd-N(pyridyl) bonds (Fig. 24.32).

Schematic representation of Ru(II)/Pd(II) coordination cage
These species preserve the weak 3MLCT luminescence of [Ru(terpy)2]2+ [156].
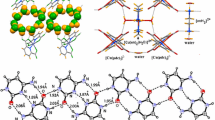
Similarly a C3-symmetric Ru(II) complex with three pendant pyridyl groups has been used as a complex-ligand with Pd(II) ions. Pd6(RuL3)8 cage has been obtained constructed by an octahedron of Pd(II) ions capped by [RuL3]2 units (Fig. 24.33). The central cavity has a volume of >5000 Å, and it encapsulates a wide range of aromatic, hydrophobic guests that are becomes protected from UV light [157].

Structural formulae of L1 and L2 ligands
Analogously, Os complexes as ligand have been used to prepare supramolecular caged in combination with cobalt or cadmium ions [158].
In the formation of this cage, the serendipity played a big role, in fact the subunit of Os(II) is formed initially in a 3 to 1 mixture of isomers mer and fac, which is exactly the combination necessary for the complete assembly of the cage. The units Os(II) tris-diimine also exhibit luminescence in the red with an appreciable lifetime such makes it possible to play the role of a good donor of photoelectrons for species encapsulated in the cavity .
3.1 Coordination Cages Based on Lanthanoids
Lanthanoids are the most common type of luminescent metal ions used in cage assembly. The systems that can be obtained with these metals have tetrahedral geometries of the type Ln4L4L4 in which a metal ion is found at each vertex and a tris chelating bridging ligand between three metals on each triangular face. Hamacek and colleagues prepared a series of Ln4L4L4 cages based on tripod ligaments such as L3–L5 containing three pyridine-dicarbonyl O,N,O,O-chelating tridentate arms, so that the Ln(III) ions are nine coordinated [159,160,161] (Fig. 24.34). Depending on the type of lanthanide /ligand combination, cages are obtained with cavities of different sizes, such as to accommodate molecules of discrete size [165] or simply series of anions [166].

Structural formulae of L3 and L13 ligands used to build-up Coordination Cages based on Lanthanoids
The typical properties of the ligand are transported into the supramolecular structure. In fact, the use of ligands in which the O,N,O-chelating units are chiral (type) allows to induce the chirality on the coordination of the metal and therefore a single diastereoisomer of the supramolecular array is obtained. The use of L6 racemic allows to obtain a mixture of the two enantiomers of the Ln4L4 tetrahedral cages as if the ligands had undergone a homochiral auto-mixing during the assembly process [162].
He, Duan and colleagues have developed a series of cages based on Ce(III)- or Ce(IV)- ions and type O,N,O-chelating bridge binders used as luminescent sensors for a wide range of guests [163,164,165,166,167,168,169,170,171,172]. The 4f-5d transition that generates Ce(III) luminescence is much more sensitive to the environment than the 4f-4f transitions associated with Eu(III) and Tb(III) luminescence and is therefore sensitive to perturbations resulting from the interactions with the hosts. In other members of the series, the oxidation state of the metal is IV, in this case the luminescence derives exclusively from the host species.
Thus, the tetrahedra of Ce4L4L4 and Ce4L6 based on face-capping (tritopic, e.g., L8) [164] and edge-bridging (ditopic, e.g., L9) [165] ligands show an increase in luminescence intensity in the presence of saccharides. This is due to the recognition based on a hydrogen-binding interaction between bound substrates and inward directed amide groups. Larger polyhedra with nuclearities based on binding agents such as L10-L12 [166, 167], show a similar luminescence response when the host is complementary to the cage cavity; thus acting as selective sensors.
Cage modifications such as the introduction of a triamine-triazine fragment in the ligand structure produce cages based on Cerium ions capable of selectively recognizing guanosine, increasing luminescence, compared to other nucleosides even in organic solvents [168]. If on the other hand a dihydropyridine amide group is introduced into the ligand skeleton, it is possible to selectively bind explosive RDX, resulting in a strong increase in luminescence compared to other explosive molecules [169].
Luminescence can be introduced into Ce4L4L4 cages by subunit of fluorescent triphenylamine in L15 ligand (Fig. 24.35); the cage obtained is an intense blue emitter [170, 171]. This species by strongly binding the organic radical 4,4,5,5,5,5-tetra-methyl-imidazolinoxyl-3-oxide is able to spin-trapping NO, and the fluorescence is substantially extinct even at 5 nM concentrations, much lower than the most commonly used EPR spectroscopic assay [172]. The same cage showed selectivity to bind tryptophan over other amino acids and was used as the basis for a luminescent test for tryptophan in serum [173].

Structural formulae of L14 and L17 ligands
The high selectivity and sensitivity of this type of sensor suggests that it can report on the progress of a reaction when a component is consumed. Some Ce4L4L4 tetrahedra of this family have been evaluated in this way; all based on a tritopic ligand type L15–L17 containing amide groups to facilitate the binding of hosts by hydrogen bonds. Reactions such as cyanosylation of aromatic aldehydes have been monitored by restoring cage luminescence [173, 174].
3.2 Coordination Cages Based on Luminescent Ligands
Luminescence in coordination cages can be introduced through the use of intrinsic luminescent subunits in binders. In these cases, however, attention must be paid to the metal ions used for assembly. This in fact must not interfere, giving phenomena of electronic transfer or non-luminescent MC states, such as to switch off the emission from excited state of organic chromophore. Ward and his collaborators have prepared a series of fluorescent cages based on ligands containing two terms pyrazolylpyridine separated by a naphthyl spacer replaced in various different geometries (L2 and L18–L20) (Figs. 24.33 and 24.36). These ligands allow the assembly of tetrahedral structures type M4L6, cubes type M8L12 cubes, and truncated tetrahedron type M12L18. The formation of π-stacked inter-ligand interactions around the periphery of the cages leads to a red shift of the luminescence typical of the formation of excimers [175,176,177].

Structural formulae of L18 and L31 ligands
It is known that tetraphenyl-ethylene units show an increase in luminescence induced by aggregation due to changes in conformation when aggregating. Stang, Huang, and colleagues have incorporated fluorophores of this type (the cages through the use of Pt(II) ions). In rigid cages, however, the rigidity of the structure forces the tetraphenyl-ethylene units to be fluorescent even at low concentrations without aggregation; when they aggregate at higher concentrations, they show tunable luminescence throughout the visible region, including white luminescence [178].
Perylene bisimide (PBI) units were used in binders by Würthner and colleagues for preparing very large tetrahedral cages (e.g., L22); edge lengths are close to 4 nm [179].The characteristic properties of PBI units are stored in cages, which therefore show very rich electrochemical and photophysical behavior. When Fe(II) is used as a metal vertex, the typical fluorescence of PBIs is switched off [179], but in a Zn(II) based cage, fluorescence centered on the ligand is preserved and can therefore be used to investigate interactions with host molecules [179]. Similarly, Nitschke and colleagues have reported M4L6 tetrahedral cages containing “BODIPY” fluorophores integrated in edge-bridging ligands (L23); the fluorescence of these species is modulated by interaction with amino acids [180]. These last cages were also decorated with coon pyrene thus obtaining a system with at the ends 12 units of pyrene and six units of BODIPY in the centers of the ligand. The only partial energy transfer efficiency between the subunits and the ability to host the blue-emitting perylene dye means that the combination of these luminophores provides an emission of white light.
A tetratopic ligand containing four units of monodentate quinoline, also used to prepare cages with lanthanum ions [181, 182], connected by amide spacers to a central aromatic nucleus forms a metallamachrocyclic group Pd3L3L3 whose cavity is selective for uridine compared to other nucleosides, always with an increase of luminescence on the bond [183].
A quinoline-based triangular binder containing L26 ligand with mono toothed sites forms a luminescent octahedral cage with Pd6L8 structure and RNA-based nucleoside sensing [183, 184].
Detecting the vapors of certain types of explosives was possible using the luminescence of prismatic cages based on Pt(II) [185,186,187,188]. These cages are prepared from a combination of multinuclear organometallic Pt/alkenes and multitopic binders, rigid, rigid, bis-, or tris-pyridine. Similar prismatic cages based on organometallic dinuclear Ru(II), with a cavity defined by the face-to-face arrangement of two triangular ligands have also been reported to act as luminescent sensors for nitroaromatics [187, 188].
Several cages made of rigid, folded, “banana-shaped” bispyridine binders have been prepared by introducing fluorescent subunits into their structure [189, 190]. The use of anthracene groups makes it possible to create a large internal cavity of about 1 nm in diameter that can accommodate a series of organic guests who are protected from the outside environment [191, 192]. If the metal ion is Pd(II), the cages are not fluorescent, and the anthracene groups only perform a structural and isolating function of the aromatic hosts [191, 192]. However, with metal ions such as Zn(II), inactive as d10, the cages show strong fluorescence with quantum yield values of up to 0.8 [193]. The analogues Ni(II) and Mn(II) were weakly luminescent, most likely, in the first case for photo-induced electronic transfer processes. The use of Cu ions makes the photophysical properties highly dependent on the solvent thanks to the degrees of coordination of the solvent molecules at the axial sites of the Cu(II) ions [194]. The anthracene fluorescence of the Hg2L4 analogue is completely switched off by photoinduced electronic transfer by encapsulation of C60 or C70 in the central cavity in MeCN [195].
Crowley and colleagues have shown how click chemistry can work on the structures of previous cages. Starting from a bent bipyridine bridge binder that brings an azide group to an external direction (L29), it is possible to introduce different substituents such as to modulate or add luminescence from external organic fluorophores to the Pd2L4 cages [196, 197].
Shionoya and colleagues have shown that by using the same gantry and metal, it is possible to obtain both luminescent and non-luminescent cages. The key lies in the use of different reports for their assembly. Using a tris-pyridyl L31 tritopic ligand and Hg(II) ions a cage with stoichiometry Hg6L8 is obtained; this cage shows strong fluorescence at 360 nm. However, when the Hg:L ratio is doubled, a nonfluorescent Hg6L4 cage is formed [198].
3.3 Luminescent Cages Based on Metallo-Porphyrin Components
There are plenty of examples of multi-component assemblies where (metal)porphyrin units have been included either in the metal’s axial site or as pendants on the periphery of the porphyrin core. This is a vast area that has been completely revised [199,200,201] due to the special relaunch of porphyrin-based assemblies to understand the photophysical processes underlying the processes of light absorption in natural photosynthesis. A beautiful review was published in 2014 [202].
The interaction of light with these supramolecular systems is not limited to fluorescence and therefore to their use as sensors.
3.4 Photochromic Coordination Cages
There are some examples of cages that, incorporating photosensitive functional groups, can be used as a basis for switching their structures or properties.
Fujita and colleagues have shown how this can be used for example to modulate the properties of the cavity, such as its hydrophobicity.
Azobenzene is a photoactive subunit particularly used for photoinduced structural changes. The introduction of this in spherical nanocages type Pd12L24 “nanosphere” has allowed under irradiation to modify the hydrophobicity of the central cavity and thus decreasing the binding of hydrophobic hosts as the derivatives of the pyrene. Heating the cage allowed the cis-azobenzene groups in the cavity to relax to the trans-forms, increasing hydrophobicity and thus restoring the bond with the guests.
In general, the conformational change associated with a photochromic group can be used to open and close a cage, resulting in modulation of absorption and release of hosts [202]. L33 ligand (Fig. 24.37) incorporates a dithenyl-ethylene group whose photoinduced opening/closing movement alters the distance between the coordinated pyridylic terms, leading to a “lengthening” and “compression” of the cage, changing the internal volume and causing a weakening of the binding of the guests in closed form [203].

Structural formulae of L32 and L38 ligands
In some cases, however, guests show useful optical or photophysical behavior, and the binding of the caged host may modulate this behavior or allow the host to be transported within his or her host [204,205,206,207,208,209,210,211].
There are now several examples of photo-induced reactions of guests tied in cages where the host cage acts as an “inert box”, providing a sterile confined space that alters the reaction pathways of the guests and then directs the reactions to provide new products [212,213,214,215,216,217,218,219].
The state of the art in this field is based on examples of a self-assembled cage capable of a series of chromophores surrounding a guest to participate in the reactions induced by the cage/host. The basic idea is to have a high local concentration of chromophores around a reactive host with a very high spatial and energetic control to obtain, for example, a photo-catalyzed reaction otherwise impossible: practically the apotheosis of supramolecular photochemistry.
Fujita [212, 215] and Ramamurthy [216, 219], have used a cage based on Pd6 that acts just like a box capable of confining and changing the course of a reaction. There are also some examples of Fujita and colleagues where the same types of cages are active participants and therefore reaction partners, along with the substrates. The first example is provided by photo-induced oxidation of an adamantane-bound host to give 1-adamantanol and 1-adamanthylhyroperoxide. Since adamantane does not absorb light, a cage with a triazine in its ligand structure allows the photoinduced transfer of electrons from adamantane to the excited state of the triazine unit. The resulting triazine radical anion is reoxidized by O2, while the adamantyl radical reacts irreversibly with O2 [220, 221]. The same photoinduced electronic transfer process involving the triazine units in the cage leads to photooxidation of other substrates as well [222, 223]. Triazine in the structure of cages, in the presence of anthracene as a host, allows the formation of a low energy absorption band that can be populated, by means of energy transfer in about 9 ps, from the excited states of the anthracene and leads to an emission of about 650 nm
Processes of energy transfer and in particular of photoinduced electron transfer have been recently published with so-called types of cages and host substrates. For example, with 4-hydroxyphenylamine as guest, irradiation of the cage causes photoinduced ET from guest to the scaffold converting the amine into a radical cation. This then loses a proton within 1 ps to the aqueous solvent to give a neutral bound radical and H3O+ [224]. In a further refinement of this methodology, using the host 9-anthracenealdehyde it generates after photoexcitation a mixture of a host radical cations (as before) and a twisted intramolecular charge transfer (TICT) state centered on the host [225].
4 Conclusions
In this chapter, we have summarized – but not exhaustive reported – the properties of coordination dendrimers, polymers and cages. The assembly of arrays of metal containing luminophores in architectures like dendrimers, polymers or coordination cages, clearly offers the way for numbers of applications. While the vast majority of research work on coordination materials is focused on their synthetic protocols, structural properties, and - in the cases of coordination cages - in guest binding capacities, the inclusion of photophysical properties or photoactivable functions offers exciting novel application opportunities.
The use of coordination polymers for the conversion of light into charge motion or electricity into light represents two complementary but important topics highly related to the solar energy conversion as well as the use of metal dendrimers as antenna systems for light harvesting or power the light induced water splitting.
The number of references in this chapter, in particular referring about molecular cages, shows how this research field is continuously growing because the use of photons– to exploit all the intrinsic electronic properties of the metal components of these materials - opens the way for innovative applications even in the case of materials, such as dendrimers, only relatively new. In fact, metal dendrimers are now considered also as good vectors for photoactivable drug delivery in cancer treatment, whereas the anticancer activity of coordination dendrimers on their own is less studied. So we hope that this chapter could act as starting point for running deeper on the marvelous world of photochemistry of multinuclear compounds.
References
(a) Tomalia, D.A., Durst, H.D.: Genealogically directed synthesis: starburst/cascade dendrimers and hyperbranched structures. Top Curr Chem 165, 193–313 (1993); (b) Fréchet, J.M.J.: Functional polymers and dendrimers: reactivity, molecular architecture, and interfacial energy. Science 263, 1710 (1994); (c) Newkome, G.R. (ed) Advances in Dendritic Macromolecules, vols 1–3. JAI Press, London (1994–1996); (d) Newkome, G.R., Moorefield, C.N., Vögtle, F. (eds) Dendritic macromolecules: concepts, syntheses, perspectives. VCH, Weinheim (1996); (e) Zeng, F., Zimmerman, S.C.: Dendrimers in supramolecular chemistry: from molecular recognition to self-assembly. Chem. Rev. 97, 1681 (1997)
(a) Serroni, S., Campagna, S., Denti, G., Juris, A., Venturi, M., Balzani, V.: Dendrimers based on metal complexes. In: Newkome, G.R. (ed) Advances in Dendritic Macromolecules, vol 3. JAI Press, London, p 61 (1996); (b) Bryce, M.R., Devonport, W.: Redox-active dendrimers, related building blocks, and oligomers. In: Newkome, G.R. (ed) Advances in Dendritic Macromolecules, vol 3. JAI Press, London, p 115 (1996); (c) Cuadrado, I., Morán, M., Losada, J., Casado, C.M., Pascual, C., Alonso, B., Lobete, F.: Organometallic dendritic macromolecules: organosilicon and organometallic entities as core or building blocks. In: Newkome, G.R. (ed) Advances in Dendritic Macromolecules, vol 3. JAI Press, London, p 151(1996)
Newkome, G.R., Moorefield, C.N., Baker, G.R., Johnson, A.L., Behera, R.K.: Alkane cascade polymers possessing micellar topology: micellanoic acid derivatives. Angew. Chem. Int. Ed. Engl. 30, 1176–1178 (1991)
Newkome, G.R., Cardullo, F., Constable, E.C., Moorefield, C.N., Thompson, A.M.W.C.: Metallomicellanols: incorporation of ruthenium(II)–2,2′: 6′,2″-terpyridine triads into cascade polymers. J. Chem. Soc. Chem. Commun. 11, 925–927 (1993)
Denti, G., Serroni, S., Campagna, S., Ricevuto, V., Balzani, V.: Directional energy transfer in a luminescent tetranuclear Ru(II) polypyridine complex that contains two different types of bridging ligands. Inorg. Chim. Acta. 182, 127–129 (1991)
Campagna, S., Denti, G., Serroni, S., Ciano, M., Juris, A., Balzani, V.: A tridecanuclear ruthenium(II)-polypyridine supramolecular species: synthesis, absorption and luminescence properties and electrochemical oxidation. Inorg. Chem. 31, 2982–2984 (1992)
Denti, G., Campagna, S., Serroni, S., Ciano, M., Balzani, V.: Decanuclear homo- and heterometallic polypyridine complexes: syntheses, absorption spectra, luminescence, electrochemical oxidation, and intercomponent energy transfer. J. Am. Chem. Soc. 114, 2944 (1992)
Alstrum-Acevedo, J.H., Brennaman, M.K., Meyer, T.J.: Chemical approaches to artificial photosynthesis. 2. Inorg. Chem. 44, 6802–6827 (2005)
Sun, L., Hammarstrom, L., Akermark, B., Styring, S.: Towards artificial photosynthesis: ruthenium–manganese chemistry for energy production. Chem. Soc. Rev. 30, 36–49 (2001)
Puntoriero, F., Sartorel, A., Orlandi, M., La Ganga, G., Serroni, S., Bonchio, M., Scandola, F., Campagna, S.: Photoinduced water oxidation using dendrimeric Ru(II) complexes as photosensitizers. Coord. Chem. Rev. 255, 2594–2601 (2011)
Arrigo, A., La Ganga, G., Nastasi, F., Serroni, S., Santoro, A., Santoni, M.-P., Galletta, M., Campagna, S., Puntoriero, F.: Artificial, molecular-based light-harvesting antenna systems made of metal dendrimers and multibodipy species. C. R. Chim. 20, 209–220 (2017)
Liao, Y.-H., Moss, J.R.: Organoruthenium dendrimers. Organometallics. 15, 4307 (1996)
Cuadrado, I., Morán, M., Casado, C.M., Alonso, B., Lobete, F., García, B., Ibisate, M., Losada, J.: Ferrocenyl-functionalized poly(propylenimine) dendrimers. Organometallics. 15, 5278 (1996)
Slany, M., Bardají, M., Casanove, M.-J., Caminade, A.-M., Majoral, J.P., Chaudret, B.: Dendrimer surface chemistry. Facile route to polyphosphines and their gold complexes. J. Am. Chem. Soc. 117, 9764 (1995)
Newkome, G.R., Cardullo, F., Constable, E.C., Moorefield, C.N., Cargill Thompson, A.M.W.: Metallomicellanols: incorporation of ruthenium(II)–2,2′: 6,2-terpyridine triads into cascade polymers. J. Chem. Soc. Chem. Commun. 7, 925 (1993)
Balzani, V., Juris, A., Venturi, M., Campagna, S.: Luminescent and redox-active polynuclear transition metal complexes. Chem. Rev. 96, 759 (1996)
Serroni, S., Denti, G., Campagna, S., Juris, A., Ciano, M., Balzani, V.: Arborols based on luminescent and redox-active transition metal complexes. Angew. Chem. Int. Ed. Engl. 31, 1493 (1992)
(a) Achar, S., Puddephatt, R.J.: Large dendrimeric organoplatinum complexes. J. Chem. Soc. Chem. Commun. 16, 1896 (1994); (b) Achar, S., Vittal, J.J., Puddephatt, R.J.: Organoplatinum dendrimers. Organometallics 15, 43–50 (1996)
Huck, W.T.S., Vanveggel, F.G.J.M., Reinhoudt, D.N.: Controlled assembly of nanosized metallodendrimers. Angew. Chem. Int. Ed. Engl. 35, 1213 (1996)
Buhlein, E., Wehner, W., Vögtle, F.: “Cascade”- and “nonskid-chain-like” syntheses of molecular cavity topologies. Synthesis. 2, 155–158 (1978)
Moors, R., Vögtle, F.: Dendrimere polyamine. Chem. Ber. 126, 2133 (1993)
(a) Wörner, C., Mülhaupt, R.: Polynitrile- and polyamine-functional poly(trimethylene imine) dendrimers. Angew. Chem. 32, 1306 (1993); (b) de Brabander-van den Berg, E.M.M., Meijer, E.W.: Poly(propylene imine) dendrimers: large-scale synthesis by hetereogeneously catalyzed hydrogenations. Angew. Chem. 32, 1308 (1993)
Cuadrado, I., Casado, C.M., Alonso, B., Moràn, M., Losada, J., Belsky, V.: Dendrimers containing organometallic moieties electronically communicated. J. Am. Chem. Soc. 119, 7613 (1997)
Serroni, S., Campagna, S., Puntoriero, F., Di Pietro, C., Loiseau, F., McClenaghan, N.D.: Dendrimers based on ruthenium(II) and osmium(II) polypyridine complexes and the approach of using complexes as ligands and complexes as metals. Chem. Soc. Rev. 30, 367 (2001)., and refs. therein
Balzani, V., Campagna, S., Denti, G., Juris, A., Serroni, S., Venturi, M.: Designing dendrimers based on transition-metal complexes. Light-harvesting properties and predetermined redox patterns. Acc. Chem. Res. 31, 26 (1998)
Hawker, C.J., Frèchet, J.M.J.: Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J. Am. Chem. Soc. 112, 7638 (1990)
Balzani, V., Juris, A., Venturi, M., Campagna, S., Serroni, S.: Luminescent and redox-active polynuclear transition metal complexes. Chem. Rev. 96, 759 (1996)
Campagna, S., Puntoriero, F., Nastasi, F., Bergamini, G., Balzani, V.: In: Balzani, V., Campagna, S. (eds.) Photochemistry and Photophysics of Coordination Compounds I, vol. 280, pp. 117–214. Springer-Verlag Berlin, Berlin (2007)
(a) Meyer, T.J.: Photochemistry of metal coordination complexes: metal to ligand charge transfer excited states. Pure Appl. Chem. 58, 1193 (1986); (b) Juris, A., Balzani, V., Barigelletti, F., Campagna, S., Belser, P., von Zelewsky, A.: Ru(II) polypyridine complexes: photophysics, photochemistry electrochemistry, and chemiluminescence. Coord. Chem. Rev. 84, 85 (1988); (c) Kalyanasundaram, K.: Photochemistry of Polypyridine and Porphyrin Complexes. Academic Press, London (1992)
Maestri, M., Balzani, V., Deuschel-Cornioley, C., von Zelewsky, A.: Adv. Photochem. 17, 1 (1992)
Bar-Haim, A., Klafter, J., Kopelman, R.: Dendrimers as controlled artificial energy antennae. J. Am. Chem. Soc. 119, 6197 (1997)
Campagna, S., Puntoriero, F., Serroni, S., Santoro, A., La Ganga, G., Galletta, M., Nastasi, F., La Mazza, E., Cancelliere, A.M.: Photo- and redox-active metal dendrimers. A journey from molecular design to applications and self-aggregated systems. Eur. J. Inorg. Chem. 35, 3887 (2018)
(a) Andersson, J., Puntoriero, F., Serroni, S., Yartsev, A., Pascher, T., Polivka, T., Campagna, S., Sundström, V.: Ultrafast singlet energy transfer competes with intersystem crossing in a multi-center transition metal polypyridine complex. Chem. Phys. Lett. 386, 336 (2004); (b) Andersson, J., Puntoriero, F., Serroni, S., Yartsev, A., Pascher, T., Polivka, T., Campagna, S., Sundström, V.: New paradigm of transition metal polypyridine complex photochemistry. Faraday Discuss. 127, 295 (2004); (c) Larsen, J., Puntoriero, F., Pascher, T., McClenaghan, N., Campagna, S., Åkesson, E., Sundström, V.: Extending the light-harvesting properties of transition-metal dendrimers ChemPhysChem. 8, 2643 (2007)
Puntoriero, F., Serroni, S., Galletta, M., Juris, A., Licciardello, A., Chiorboli, C., Campagna, S., Scandola, F.: A new Heptanuclear dendritic ruthenium(II) complex featuring photoinduced energy transfer across high-energy subunits. ChemPhysChem. 6, 129 (2005)
(a) Haga, M.-A., Ali, M.M., Arakawa, R.: Proton-induced switching of electron transfer pathways in dendrimer-type tetranuclear RuOs3 complexes, Angew. Chem. Int. Ed. Engl. 35, 76 (1996); (b) Latterini, L., Schweitzer, G., De Schryver, F.C., Moucheron, C., Kirsch-De Mesmaeker, A.: Femtosecond transient dynamics of a heptametallic HAT-ruthenium(II) complex. A photophysical study. Chem. Phys. Lett. 267, 281 (1997); (c) Latterini, L., Pourtois, G., Moucheron, C., Lazzaroni, R., Brédas, J.-L., Kirsch-De Mesmaeker, A., De Schryver, F.C.: STM imaging of a heptanuclear ruthenium(II) dendrimer, mono-add layer on graphite, Chem. Eur. J., 6, 1331 (2000); (d) Troian-Gautier, L., Moucheron, C.: Ruthenium II complexes bearing fused polycyclic ligands: from fundamental aspects to potential applications, Molecules 19, 5028 (2014)
La Mazza, E., Puntoriero, F., Nastasi, F., Laramée-Milette, B., Hananand, G.S., Campagna, S.: A heptanuclear light-harvesting metal-based antenna dendrimer with six Ru(II)-based chromophores directly powering a single Os(II)-based energy trap. Dalton Trans. 45, 19238–19241 (2016)
(a) Haga, M.-A., Ali, M.M., Arakawa, R.: Proton-induced switching of electron transfer pathways in dendrimer-type tetranuclear RuOs3 complexes. Angew. Chem. Int. Ed. Engl. 35, 76 (1996); (b) Latterini, L., Schweitzer, G., De Schryver, F.C., Moucheron, C., Kirsch-De Mesmaeker, A.: Femtosecond transient dynamics of a heptametallic HAT–ruthenium(II) complex. A photophysical study Chem. Phys. Lett., 267, 281 (1997); (c) Latterini, L., Pourtois, G., Moucheron, C., Lazzaroni, R., Brédas, J.-L., Kirsch-De Mesmaeker, A., De Schryver, F.C.: STM imaging of a heptanuclear ruthenium(II) dendrimer, mono-add layer on graphite. Chem. Eur. J. 6, 1331 (2000); (d) Troian-Gautier, L., Moucheron, C.: RutheniumII complexes bearing fused polycyclic ligands: from fundamental aspects to potential applications. Molecules 19, 5028 (2014)
Arrigo, A., Puntoriero, F., La Ganga, G., Campagna, S., Burian, M., Bernstorff, S., Amenitsch, H.: Aggregation-induced energy transfer in a decanuclear Os(II)/Ru(II) polypyridine light- harvesting antenna dendrimer. Chem. 3, 494 (2017)
Sommovigo, M., Denti, G., Serroni, S., Campagna, S., Mingazzini, C., Mariotti, C., Juris, A.: Polynuclear polypyridine complexes incorporating Ru(II), Os(II), and Pt(II): decanuclear dendrimeric antennas. Inorg. Chem. 40, 3318 (2001)
McClenaghan, N.D., Loiseau, F., Puntoriero, F., Serroni, S., Campagna, S.: Light-harvesting metal dendrimers appended with additional organic chromophores: a tetranuclear heterometallic first-generation dendrimer exhibiting unusual absorption features. Chem. Commun. 24, 2634–2635 (2001)
Serroni, S., Campagna, S., Juris, A., Venturi, M., Balzani, V., Denti, G.: Polyether arborols mounted on a luminescent and redox-active ruthenium (II)-polypyridine core. Gazz. Chim Ital. 124, 423–427 (1994)
Sadamoto, R., Tomioka, N., Aida, T.: Photoinduced electron transfer reactions through dendrimer architecture. J. Am. Chem. Soc. 118, 3978 (1996)
Issberner, J., Vögtle, F., De Cola, L., Balzani, V.: Dendritic bipyridine ligands and their tris(bipyridine)ruthenium(II) chelates—syntheses, absorption spectra, and photophysical properties. Chem. Eur. J. 3, 706 (1997)
Plevoets, M., Vögtle, F., de Cola, L., Balzani, V.: Supramolecular dendrimers with a [Ru(bpy)3]2+ core and naphthyl peripheral units. New J. Chem. 23, 63–69 (1999)
Kawa, M., Fréchet, J.M.J.: Self - assembled lanthanide - cored dendrimer complexes: enhancement of the luminescence properties of lanthanide ions through site - isolation and antenna effects. Chem. Mater. 10, 286–296 (1998)
Vicinelli, V., Ceroni, P., Maestri, M., Balzani, V., Gorka, M., Vögtle, F.: Luminescent lanthanide ions hosted in a fluorescent polylysin dendrimer. Antenna - like sensitization of visible and near - infrared emission. J. Am. Chem. Soc. 124, 6461–6468 (2002)
Ma, H., Jen, A.K.Y.: Functional dendrimers for nonlinear optics. Adv. Mater. 13, 1201–1205 (2001)
Andraud, C., Fortrie, R., Barsu, C., Stephan, O., Chermette, H., Baldeck, P.L.: Excitonically coupled oligomers and dendrimers for two - photon absorption. Adv. Polym. Sci. 214, 149–203 (2008)
Powell, C.E., Morrall, J.P., Ward, S.A., Cifuentes, M.P., Notaras, E.G.A., Samoc, M., Humphrey, M.G.: Dispersion of the third - order nonlinear optical properties of an organometallic dendrimer. J. Am. Chem. Soc. 126, 12234–12235 (2004)
(a) Cifuentes, M.P., Powell, C.E., Morrall, J.P., McDonagh, A.M., Lucas, N.T., Humphrey, M.G., Samoc, M., Houbrechts, S., Asselberghs, I., Clays, K., Persoons, A., Isoshima, T.: Electrochemical, spectroelectrochemical, and molecular quadratic and cubic nonlinear optical properties of alkynylruthenium dendrimers. J. Am. Chem. Soc. 128, 10819–10832 (2006); (b) Samoc, M., Morrall, J.P., Dalton, G.T., Cifuentes, M.P., Humphrey, M.G.: Two -photon and three - photon absorption in an organometallic dendrimer. Angew. Chem. Int. Ed. 46, 731–733 (2007)
Yang, X., Jiang, Q., Shi, P.: Two-photon absorptive multinuclear complexes. Progress in Chemistry. 30(8), 1172–1185 (2018)
Simpson, P.V., Watson, L.A., Barlow, A., Wang, G., Cifuentes, M.P., Humphrey, M.G.: Record multiphoton absorption cross-sections by dendrimer organometalation. Angew. Chem. Int. Ed. 55, 2387–2391 (2016)
Yang, J., Cho, S., Yoo, H., Park, J., Li, W.-S., Aida, T., Kim, D.: Control of molecular structures and photophysical properties of zinc(II) porphyrin dendrimers using bidentate guests: utilization of flexible dendrimer structures as a controllable mold. J. Phys. Chem. A. 112, 6869–6876 (2008)
Imahori, H.: Giant multiporphyrin arrays as artificial light-harvesting antennas. J. Phys. Chem. B. 108, 6130–6143 (2004)
Yang, J., Yoon, M.-C., Yoo, H., Kim, P., Kim, D.: Excitation energy transfer in multiporphyrin arrays with cyclic architectures: towards artificial light-harvesting antenna complexes. Chem. Soc. Rev. 41, 4808–4826 (2012)
Benites, M.R., Johnson, T.E., Weghorn, S., Yu, L., Rao, P.D., Diers, J.R., Yang, S.I., Kirmaier, C., Bocian, D.F., Holten, D., Lindsey, J.S.: Synthesis and properties of weakly coupled dendrimeric multiporphyrin light-harvesting arrays and hole-storage reservoirs. J. Mater. Chem. 12, 65–80 (2002)
Cho, S., Li, W.-S., Yoon, M.-C., Ahn, T.K., Jiang, D.-L., Kim, J., Aida, T., Kim, D.: Relationship between incoherent excitation energy migration processes and molecular structures in zinc(II) porphyrin dendrimers. Chem. Eur. J. 12, 7576 (2006)
(a) Choi, M.-S., Aida, T., Yamazaki, T., Yamazaki, I.: A large dendritic multiporphyrin array as a mimic of the bacterial light-harvesting antenna complex: molecular design of an efficient energy funnel for visible photons. Angew. Chem. Int. Ed. 40, 3194–3198 (2001); (b) Choi, M.-S., Yamazaki, T., Yamazaki, I., Aida, T.: Bioinspired molecular design of light-harvesting multiporphyrin arrays. Angew. Chem. Int. Ed. 43, 150–158 (2004)
Lee, H., Jeong, Y.-H., Kim, J.-H., Kim, I., Lee, E., Jang, W.-D.: Supramolecular coordination polymer formed from artificial light-harvesting dendrimer. J. Am. Chem. Soc. 137(38), 12394–12399 (2015)
Mede, T., Jäger, M., Schubert, U.S.: “Chemistry-on-the-complex”: functional RuII polypyridyl-type sensitizers as divergent building blocks. Chem. Soc. Rev. 47, 7577 (2018)
Adronov, A., Fréchet, J.M.J.: Light-harvesting dendrimers. Chem. Commun. (18), 1701 (2000)
Newkome, G.R., Moorefield, C., Vögtle, F.: Dendrimers and Dendrons. Wiley-VCH, Weinheim (2001)
Constable, E.C.: Metallodendrimers: metal ions as supramolecular glue. Chem. Commun. 12, 1073 (1997) and references therein
Borje, A., Kothe, O., Juris, A.: A new bridging ligand for the synthesis of luminescent polynuclear Ru(II) and Os(II) polypyridine complexes. New J. Chem. 25, 191 (2001)
Constable, E.C., Handel, R.W., Housecroft, C.E., Morales, A.F., Ventura, B., Flamigni, L., Barigelletti, F.: Metal-directed synthesis and photophysical studies of trinuclear V-shaped and pentanuclear X-shaped ruthenium and osmium metallorods and metallostars based upon 4′-(3,5-dihydroxyphenyl)-2,2′:6′,2″-terpyridine divergent units. Chem. Eur. J. 11, 4024–4034 (2005)
Constable, E.C., Housecroft, C.E., Neuburger, M., Poleschak, I., Zehnder, M.: Functionalised 2,2′-bipyridine ligands for the preparation of metallostars: X-ray structures of free ligands and preparation of copper(I) and silver(I) complexes. Polyhedron. 22, 93–108 (2003)
Bozic-Weber, B., Constable, E.C., Figgemeier, E., Housecroft, C.E., Kylberg, W.: Evaluation of polynuclear dendrons as photosensitizers for dye-sensitized solar cells. Energy Environ. Sci. 2, 299–305 (2009)
Constable, E.C., Harverson, P., Housecroft, C.E., Nordlander, E., Olsson, J.: Conventional and metal-directed synthesis of homodinuclear and heterotrinuclear complexes of homoditopic and heteroditopic ligands incorporating bpy and tpy metal-binding domains. Polyhedron. 25, 437–458 (2006)
Poddutoori, P.K., Poddutoori, P., Maiya, B.G.: Synthesis, spectroscopy and photochemistry of dyads and triads with porphyrins and bis(terpyridine)ruthenium(II) complex. J. Porphyrins Phthalocyanines. 10, 1049–1060 (2006)
Bodige, S., Torres, A.S., Maloney, D.J., Tate, D., Kinsel, G.R., Walker, A.K., MacDonnell, F.M.: First-generation chiral metallodendrimers: stereoselective synthesis of rigid D3-symmetric tetranuclear ruthenium complexes. J. Am. Chem. Soc. 119, 10364 (1997)
Kim, M.-J., MacDonnell, F.M., Gimon-Kinsel, M.E., Du Bois, T., Asgharian, N., Griener, J.C.: Global chirality in rigid decametallic ruthenium dendrimers. Angew. Chem. Int. Ed. 39, 615 (2000)
Knuckeyand, K.J., Williams, J.A.G.: Photon funnels for one-way energy transfer: multimetallic assemblies incorporating cyclometallated iridium or rhodium units accessed by sequential cross-coupling and bromination. Eur. J. Inorg. Chem. 44, 5205–5214 (2017)
Winter, A., Schubert, U.S.: Synthesis and characterization of metallo-supramolecular polymers. Chem. Soc. Rev. 45, 5311 (2016)
Kenawy, E.-R., Worley, S.D., Broughton, R.: The chemistry and applications of antimicrobial polymers: a state-of-the-art review. Biomacromolecules. 8, 1359 (2007)
Montemor, M.F.: Functional and smart coatings for corrosion protection: a review of recent advances. Surf. Coat. Technol. 258, 17–37 (2014)
Hager, M.D., Greil, P., Leyens, C., van der Zwaag, S., Schubert, U.S.: Self-healing materials. Adv. Mater. 22, 5424 (2010)
Hu, J., Zhu, Y., Huang, H., Lu, J.: Recent advances in shape–memory polymers: structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 37, 1720 (2012)
Brannon-Peppas, L., Blanchette, J.O.: Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 64, 206 (2012)
Rosler, A., Vandermeulen, G.W.M., Klok, H.-A.: Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 64, 270–279 (2012)
Pack, D.W., Hoffman, A.S., Pun, S., Stayton, P.S.: Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 4, 581–593 (2005)
Ganta, S., Devalapally, H., Shahiwala, A., Amiji, M.: A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release. 126, 187 (2008)
Wolfbeis, O.S.: An overview of nanoparticles commonly used in fluorescent bioimaging. Chem. Soc. Rev. 44, 4743 (2015)
Luckachan, G.E., Pillai, C.K.S.: Biodegradable polymers- a review on recent trends and emerging perspectives. J. Polym. Environ. 19, 637 (2011)
Li, G., Zhu, R., Yang, Y.: Polymer solar cells. Nat. Photonics. 6, 153 (2012)
Nyholm, L., Nystroem, G., Mihranyan, A., Strmme, M.: Toward flexible polymer and paper-based energy storage devices. Adv. Mater. 23, 3751 (2011)
Janoschka, T., Hager, M.D., Schubert, U.S.: Powering up the future: radical polymers for battery applications. Adv. Mater. 24, 6397 (2012)
Xiang, J., Ho, C.-L., Wong, W.-Y.: Metallopolymers for energy production, storage and conservation. Polym. Chem. 6, 6905 (2015)
Facchetti, A.: π-Conjugated polymers for organic electronics and photovoltaic cell applications. Chem. Mater. 23, 733 (2010)
Grimsdale, A.C., Leok Chan, K., Martin, R.E., Jokisz, P.G., Holmes, A.B..: Synthesis of light-emitting conjugated polymers for applications in electroluminescent devices. Chem. Rev. 109, 897 (2009)
Rehahn, M.: Organic/inorganic hybrid polymers. Acta Polym. 49, 201 (1998)
Ciardelli, F., Tsuchida, E., Wöhrle, D. (eds.): Macromolecule-Metal Complexes. Springer Verlag, Berlin (1996)
Manners, I.: Putting metals into polymers. Science. 294, 1664 (2001)
Ho, C.-L., Wong, W.-Y.: Metal-containing polymers: facile tuning of photophysical traits and emerging applications in organic electronics and photonics. Coord. Chem. Rev. 255, 2469–2502 (2011)
Winter, A., Friebe, C., Chiper, M., Hager, M.D., Schubert, U.S.: Self-assembly of π-conjugated bis(terpyridine) ligands with zinc(II) ions: new metallo-supramolecular materials for optoelectronic applications. J. Polym. Sci. A Polym. Chem. 47, 4083 (2009)
Morisige, K.: Metal complexes of aromatic schiff base compounds: Part I. The fluorescence properties of aluminium and gallium complexes of aromatic schiff bases and their use in fluorimetry. Anal. Chim. Acta. 72, 295 (1974)
Kwok, C.-C., Yu, S.-C., Sham, I.H.T., Che, C.-M.: Self-assembled zinc(II) Schiff base polymers for applications in polymer light-emitting devices. Chem. Commun. 23, 2758 (2004)
Peng, Q., Xie, M., Huang, Y., Lu, Z., Cao, Y.: Novel supramolecular polymers based on zinc-salen chromophores for efficient light-emitting diodes. Macromol. Chem. Phys. 206, 2373 (2005)
Constable, E.C.: Towards helical coordination polymers: molecular wires in chiral coats. Macromol. Symp. 98, 503 (1995)
Dobrawa, R., Würthner, F.: Photoluminescent supramolecular polymers: metal-ion directed polymerization of terpyridine-functionalized perylene bisimide dyes. Chem. Commun. 17, 1878 (2002)
Yu, S.-Y., Kwok, C.-C., Chan, W.-K., Che, C.-M.: Self-assembled electroluminescent polymers derived from terpyridine-based moieties. Adv. Mater. 15, 1643 (2003)
Winter, A., Friebe, C., Chiper, M., Hager, M.D., Schubert, U.S.: Self-assembly of π-conjugated bis(terpyridine) ligands with zinc(II) ions: new metallosupramolecular materials for optoelectronic applications. J. Polym. Sci. A Polym. Chem. 47, 4083 (2009)
Schlütter, F., Wild, A., Winter, A., Hager, M.D., Baumgaertel, A., Friebe, C., Schubert, U.S.: Synthesis and characterization of new self-assembled metallo-polymers containing electron-withdrawing and electron-donating Bis(terpyridine) zinc(II) moieties. Macromolecules. 43, 2759 (2010)
Chen, Y.-Y., Lin, H.-C.: Synthesis and characterization of light-emitting main-chain metallo-polymers containing bis-terpyridyl ligands with various lateral substituents. J. Polym. Sci. A Polym. Chem. 45, 3243 (2007)
Meyers, A., South, C., Weck, M.: Design, synthesis, characterization, and fluorescent studies of the first zinc-quinolate polymer. Chem. Commun. 10, 1176 (2004)
Hayashida, N., Yamamoto, T.: Synthesis of Ru- and Os-complexes of π-conjugated oligomers of 2,2′-bipyridine and 5,5′-bipyrimidine. Optical properties and catalytic activity for photoevolution of H2 from aqueous media. Bull. Chem. Soc. Jpn. 72, 1153 (1999)
Yu, S.C., Gong, X., Chan, W.K.: Synthesis and characterization of poly(benzobisoxazole)s and poly(benzobisthiazole)s with 2,2’-bipyridyl units in the backbone. Macromolecules. 31, 5639 (1998)
Peng, Z., Yu, L.: Synthesis of conjugated polymers containing ionic transition metal complexes. J. Am. Chem. Soc. 118, 3777 (1996)
Wang, Q., Yu, L.: Conjugated polymers containing mixed-ligand ruthenium(II) complexes. Synthesis, characterization, and investigation of photoconductive properties. J. Am. Chem. Soc. 122, 11806 (2000)
Peng, Z., Gharavi, A.R., Yu, L.: Synthesis and characterization of photorefractive polymers containing transition metal complexes as photosensitizer. J. Am. Chem. Soc. 119, 4622 (1997)
Ng, P.K., Gong, X., Chan, S.H., Lam, L.S.M., Chan, W.K.: The role of ruthenium and rhenium diimine complexes in conjugated polymers that exhibit interesting opto-electronic properties. Chem. Eur. J. 7, 4358 (2001)
Walters, K.A., Trouillet, L., Guillerez, S., Schanze, K.S.: Photophysics and electron transfer in poly(3-octylthiophene) alternating with Ru(II)- and Os(II)−bipyridine complexes. Inorg. Chem. 39, 5496 (2000)
Yu, S.C., Hou, S., Chan, W.K.: Synthesis, metal complex formation, and electronic properties of a novel conjugate polymer with a tridentate 2,6-bis(benzimidazol-2-yl)pyridine ligand. Macromolecules. 32, 5251 (1999)
Man, K.Y.K., Wong, H.L., Chan, W.K.: Use of a ruthenium-containing conjugated polymer as a photosensitizer in photovoltaic devices fabricated by a layer-by-layer deposition process. Langmuir. 22, 3368 (2006)
Ng, W.Y., Gong, X., Chan, W.K.: Electronic and light-emitting properties of some polyimides based on bis(2,2′:6′,2″-terpyridine) ruthenium(II) complex. Chem. Mater. 11, 1165 (1999)
Yu, S.C., Hou, S.J., Chan, W.K.: Synthesis and properties of polyamides and polyesters on the basis of 2,2′-bipyridine-5,5′-dicarboxylic acid and the corresponding polymer−ruthenium complexes. Macromolecules. 33, 3259 (2000)
Wong, C.T., Chen, W.K.: Yellow light–emitting poly(phenylenevinylene) incorporated with pendant ruthenium bipyridine and terpyridine complexes. Adv. Mater. 11, 455 (1999)
Liu, Y., Wang, Z., Zhang, X.: Characterization of supramolecular polymers. Chem. Soc. Rev. 41, 5922 (2012)
Langecker, J., Rehahn, M.: Iridium-functionalized polyfluorenes: advantages and limitations of the Suzuki and Yamamoto approaches. Macromol. Chem. Phys. 209, 258 (2008)
Ito, T., Suzuki, S., Kido, J.: Synthesis of polymer-iridium complex and its electroluminescent characteristics. Polym. Adv. Technol. 16, 480 (2005)
Zhang, K., Chen, Z., Yang, C., Gong, S., Qin, J., Cao, Y.: Saturated red-emitting electrophosphorescent polymers with iridium coordinating to β-diketonate units in the main chain. Macromol. Rapid Commun. 27, 1926 (2006)
Park, M.-J., Kwak, J., Lee, J., Jung, I.H., Kong, H., Lee, C., Hwang, D.-H., Shim, H.-K.: Single chain white-light-emitting polyfluorene copolymers containing iridium complex coordinated on the main chain. Macromolecules. 43, 1379 (2010)
Zhang, K., Chen, Z., Yang, C., Zou, Y., Gong, S., Tao, Y., Qin, J., Cao, Y.: Iridium complexes embedded into and end-capped onto phosphorescent polymers: optimizing PLED performance and structure–property relationships. J. Mater. Chem. 18, 3366 (2008)
Yang, W., Zhen, H.Y., Jiang, C.Y., Su, L.J., Jiang, J.X., Shi, H.H., Cao, Y.: Synthesis of electrophosphorescent polymers based on para-phenylenes with iridium complexes. Synth. Met. 153, 189 (2005)
Guan, R., Zu, Y., Ying, L., Yang, W., Wu, H., Chen, Q., Cao, Y.: Novel green-light-emitting hyperbranched polymers with iridium complex as core and 3,6-carbazole-co-2,6-pyridine unit as branch. J. Mater. Chem. 19, 531 (2009)
Chen, X., Liao, J.-L., Liang, Y., Ahmed, M.O., Tseng, H.-E., Chen, S.A.: High-efficiency red-light emission from polyfluorenes grafted with cyclometalated iridium complexes and charge transport moiety. J. Am. Chem. Soc. 125, 636 (2003)
Mei, C., Ding, J., Yao, B., Cheng, Y., Xie, Z., Geng, Y., Wang, L.: Synthesis and characterization of white-light-emitting polyfluorenes containing orange phosphorescent moieties in the side chain. J. Polym. Sci. A Polym. Chem. 45, 1746–1757 (2007)
Fujikura, Y., Sonogashira, K., Hagihara, N.: Preparation and uv spectra of some oligomer-complexes composed of platinum group metals and conjugated poly-yne systems. Chem. Lett. 4, 1067–1070 (1975)
Takahashi, S., Kariya, M., Yatake, T., Sonogashira, K., Hagihara, N.: Studies of poly-yne polymers containing transition metals in the main chain. 2. Synthesis of poly[trans-bis(tri-n-butylphosphine)platinum 1,4-butadiynediyl] and evidence of a rodlike structure. Macromolecules. 11, 1063 (1978)
Sonogashira, K., Takahashi, S., Hagihara, N.: A new extended chain polymer, poly[trans-bis(tri-n-butylphosphine)platinum 1,4-butadiynediyl]. Macromolecules. 10, 879 (1977)
Markwell, R.D., Butler, I.S., Kakkar, A.K., Khan, M.S., Al-Zakwani, Z.H., Lewis, J.: Vibrational spectroscopic investigation of rigid-rod platinum σ-acetylide polymers containing variable acetylenic microstructures. Organometallics. 15, 2331 (1996)
Lewis, J., Khan, M.S., Johnson, B.F.G., Marder, T.B., Fyfe, H.B., Wittmann, F., Friend, R.H., Dray, A.E.: Di-, tri-, pseudo-di- and pseudo-tetra-acetylenic polymers of platinum: synthesis, characterization and optical spectra. J. Organomet. Chem. 425, 165–176 (1992)
Chawdhury, N., Köhler, A., Friend, R.H., Wong, W.-Y., Lewis, J., Younus, M., Raithby, P.R., Corcoran, T.C., Al-Mandhary, M.R.A., Khan, M.S.: Evolution of lowest singlet and triplet excited states with number of thienyl rings in platinum poly-ynes. J. Chem. Phys. 110, 4963 (1999)
Bunten, K.A., Kakkar, A.K.: Synthesis of pyridine/pyridinium-based alkynyl monomers, oligomers and polymers: enhancing conjugation by pyridine N-quaternization. J. Mater. Chem. 5, 2041–2043 (1995)
Khan, M.S., Al-Mandhary, M.R.A., Al-Suti, M.K., Hisahm, A.K., Raithby, P.R., Ahrens, B., Mahon, M.F., Male, L., Marseglia, E.A., Tedesco, E., Friend, R.H., Köhler, A., Feeder, N., Teat, S.J.: Structural characterisation of a series of acetylide-functionalised oligopyridines and the synthesis, characterisation and optical spectroscopy of platinum di-ynes and poly-ynes containing oligopyridyl linker groups in the backbone. J. Chem. Soc. Dalton Trans. 0, 1358–1368 (2002)
Chawdhury, N., Köhler, A., Friend, R.H., Younus, M., Long, N.J., Raithby, P.R., Lewis, J.: Synthesis and electronic structure of platinum-containing poly-ynes with aromatic and heteroaromatic rings. Macromolecules. 31, 722 (1998)
Wong, W.-Y., Wong, C.-K., Lu, G.-L., Cheah, K.-W., Shi, J.-X., Lin, Z.: Synthesis, structures and optical spectroscopy of photoluminescent platinum-linked poly(silylacetylenes). J. Chem. Soc. Dalton Trans. 24, 4587 (2002)
Wong, W.-Y., Wong, C.-K., Lu, G.-L., Lee, A.W.-M., Cheah, K.-W., Shi, J.-X.: Triplet emission in platinum-containing poly(alkynylsilanes). Macromolecules. 36, 983 (2003)
Wong, W.-Y., Poon, S.-Y., Shi, J.-X., Cheah, K.-W.: Synthesis and light-emitting properties of platinum-containing oligoynes and polyynes derived from oligo(fluorenyleneethynylenesilylene)s J. Polym. Sci. A: Polym. Chem. 44, 4804 (2006)
Wong, W.-Y., Poon, S.-Y., Shi, J.-X., Cheah, K.-W.: Synthesis, optical properties, and photoluminescence of organometallic acetylide polymers of platinum functionalized with Si and Ge-bridged bis(3,6-diethynyl-9-butylcarbazole). J. Inorg. Organomet. Polym. Mater. 17, 189 (2007)
Wong, W.-Y., Lu, G.-L., Choi, K.-H., Shi, J.-X.: Synthesis and electronic properties of new photoluminescent platinum-containing polyynes with 9,9-dihexylfluorene and 9-butylcarbazole units. Macromolecules. 25, 3506 (2002)
Zhou, G.-J., Wong, W.-Y., Cui, D., Ye, C.: Anhydrous praseodymium salts in the ionic liquid [bmpyr][Tf2N]: structural and optical properties of [bmpyr]4[PrI6][Tf2N] and [bmyr]2[Pr(Tf2N)5]. Chem. Mater. 17, 5209 (2005)
Lewis, J., Raithby, P.R., Wong, W.-Y.: Synthesis of new bis(acetylide)-substituted fluorene derivatives and their bimetallic and polymeric complexes. J. Organomet. Chem. 219, 556 (1998)
Wong, W.-Y., Choi, K.-H., Lu, G.-L., Shi, J.-X.: Synthesis, redox and optical properties of low-bandgap platinum(II) polyynes with 9-dicyanomethylene-substituted fluorene acceptors. Macromol. Rapid Commun. 22, 461 (2001)
Wong, W.-Y., Wong, W.-K., Raithby, P.R.: Synthesis, characterisation and properties of platinum(II) acetylide complexes of ferrocenylfluorene: novel soluble donor–acceptor heterometallic materials. J. Chem. Soc. Dalton Trans. 16, 2761 (1998)
Liu, L., Wong, W.-Y., Poon, S.-Y., Shi, J.-X., Cheah, K.-W., Lin, Z.: Crystalline carbon hollow spheres, crystalline carbon−SnO2 hollow spheres, and crystalline SnO2 hollow spheres: synthesis and performance in reversible Li-Ion storage. Chem. Mater. 18, 1369 (2006)
Ward, M.D.: Photophysical properties of coordination cages and their host/guest assemblies. Compr. Supramol. Chem. II. 6, 357 (2017)
Saalfrank, R.W.W., Stark, A., Peters, K., von Schnering, H.G.: The first “adamantoid” alkaline earth metal chelate complex: synthesis, structure, and reactivity. Angew. Chem. Int. Ed. 27, 851–853 (1988)
Saalfrank, R.W., Stark, A., Bremer, M., Hummel, H.-U.: Formation of tetranuclear chelate(4-) ions of divalent metals (Mn, Co, Ni) with idealized T symmetry by spontaneous self-assembly. Angew. Chem. Int. Ed. Ed. Engl. 29, 311–314 (1990)
Harris, K., Fujita, D., Fujita, M.: Giant hollow MnL2n spherical complexes: structure, functionalisation and applications. Chem. Commun. 49, 6703–6712 (2013)
Sun, Q.-F., Iwasa, J., Ogawa, D., Ishido, Y., Sato, S., Ozeki, T., Sei, Y., Yamaguchi, K., Fujita, M.: Self-assembled M24L48 polyhedra and their sharp structural switch upon subtle ligand variation. Science. 328, 1144–1147 (2010)
Alvarez, S.: Polyhedra in (inorganic) chemistry. Dalton Trans. 13, 2209–2233 (2005)
Cook, T.R., Stang, P.J.: Recent developments in the preparation and chemistry of metallacycles and metallacages via coordination. Chem. Rev. 115, 7001–7045 (2015)
Beissel, T., Powers, R.E., Raymond, K.N.: Symmetry-based metal complex cluster formation. Angew. Chem. Int. Ed. 35, 1084–1086 (1996)
Olenyuk, B., Levin, M.D., Whiteford, J.A., Shield, J.E., Stang, P.J.: Self-assembly of nanoscopic dodecahedra from 50 predesigned components. J. Am. Chem. Soc. 121, 10434–10435 (1999)
Chepelin, O., Ujma, J., Wu, X., Slawin, A.M.Z., Pitak, M.B., Coles, S.J., Michel, J., Jones, A.C.C., Barran, P.E., Lusby, P.J.J.: Luminescent, enantiopure, phenylatopyridine iridium-based coordination capsules. J. Am. Chem. Soc. 134, 19334–19337 (2012)
Yang, J., Bhadbhade, M., Donald, W.A., Iranmanesh, H., Moore, E.G., Yan, H., Beves, J.E.: Self-assembled supramolecular cages containing ruthenium(II) polypyridyl complexes. Chem. Commun. 51, 4465–4468 (2015)
Li, K., Zhang, L.-Y., Yan, C., Wei, S.-C., Pan, M., Zhang, L., Su, C.-Y.: Stepwise assembly of Pd6(RuL3)8 nanoscale rhombododecahedral metal–organic cages via metalloligand strategy for guest trapping and protection. J. Am. Chem. Soc. 136, 4456–4459 (2014)
Wragg, A.B.., Metherell, A.J., Cullen, W., Ward, M.D.: Stepwise assembly of mixed-metal coordination cages containing both kinetically inert and kinetically labile metal ions: introduction of metal-centred redox and photophysical activity at specific sites. Dalton Trans. 44, 17939–17949 (2015)
Hamacek, J., Bernardinelli, G., Filinchuk, Y.: Tetrahedral assembly with lanthanides: toward discrete polynuclear complexes. Eur. J. Inorg. Chem. 22, 3419–3422 (2008)
El Aroussi, B., Guénée, L., Pal, P., Hamacek, J.: Lanthanide-mediated supramolecular cages and host–guest interactions. Inorg. Chem. 50, 8588–8597 (2011)
Hamacek, J., Poggiali, D., Zebret, S., El Aroussi, B., Schneider, M.W., Mastalerz, M.: Building large supramolecular nanocapsules with europium cations. Chem. Commun. 48, 1281–1283 (2012)
Yan, L.-L., Tan, C.-H., Zhang, G.-L., Zhou, L.-P., Bünzli, J.-C., Sun, Q.-F.: Stereocontrolled self-assembly and self-sorting of luminescent europium tetrahedral cages. J. Am. Chem. Soc. 137, 8550–8555 (2015)
Liu, Y., Lin, Z., He, C., Zhao, L., Duan, C.: A symmetry-controlled and face-driven approach for the assembly of cerium-based molecular polyhedral. Dalton Trans. 39, 11122–11125 (2010)
Zhang, J., He, C., Duan, C.: Self-assembled cerium-based metal–organic tetrahedrons for selective recognition of natural saccharides. Inorg. Chem. Commun. 49, 140–142 (2014)
Liu, Y., Wu, X., He, C., Jiao, Y., Duan, C.: Self-assembly of cerium-based metal–organic tetrahedrons for size-selectively luminescent sensing natural saccharides. Chem. Commun. 48, 7554–7556 (2009)
Jiao, Y., Zhang, J., Zhang, L., Lin, Z., He, C., Duan, C.: Metal–organic polyhedra containing 36 and 24 folds of amide groups for selective luminescent recognition of natural disaccharides. Chem. Commun. 48, 6022 (2012)
Zhao, L., Qu, S., Zhang, R., Duan, C.: Face-driven octanuclear cerium(IV) luminescence polyhedra: synthesis and luminescent sensing natural saccharides. Chem. Commun. 47, 9387–9389 (2011)
Zhang, J., He, C., Duan, C.: Cerium-based metal–organic tetrahedron for selective sensing of ribonucleosides through the cooperation of hydrogen bonding interaction. Inorg. Chem. Commun. 54, 41–44 (2015)
Zhao, L., Chu, Y., He, C., Duan, C.: Fluorescent detection of RDX within DHPA-containing metal–organic polyhedral. Chem. Commun. 50, 3467–3469 (2014)
Wang, J., He, C., Wu, P., Wang, J., Duan, C.: An amide-containing metal–organic tetrahedron responding to a spin-trapping reaction in a fluorescent enhancement manner for biological imaging of NO in living cells. J. Am. Chem. Soc. 133, 12402–12405 (2011)
He, C., Wang, J., Wu, P., Jia, L., Bai, Y., Zhang, Z., Duan, C.: Fluorescent differentiation and quantificational detection of free tryptophan in serum within a confined metal–organic tetrahedron. Chem. Commun. 48, 11880–11882 (2012)
Jiao, Y., Wang, J., Wu, P., Zhao, L., He, C., Zhang, J., Duan, C.: Cerium-based M4L4 Tetrahedra as molecular flasks for selective reaction prompting and luminescent reaction tracing. Chem. Eur. J. 20, 2224–2231 (2014)
Zhang, J., Yu, H., Zhang, C., He, C., Duan, C.: Cerium-based M4L4 tetrahedrons containing hydrogen bond groups as functional molecular flasks for selective reaction prompting. New J. Chem. 38, 3137–3145 (2014)
Wang, P., Ma, J.-P., Dong, Y.-B.: Guest-driven luminescence: lanthanide-based host–guest systems with bimodal emissive properties based on a guest-driven approach. Chem. Eur. J. 15, 10432–10445 (2009)
Al-Rasbi, N.K., Sabatini, C., Barigelletti, F., Ward, M.D.: Red-shifted luminescence from naphthalene-containing ligands due to π-stacking in self-assembled coordination cages. Dalton Trans. 40, 4768–4772 (2006)
Tidmarsh, I.S., Faust, T.B., Adams, H., Harding, L.P., Russo, L., Clegg, W., Ward, M.D.: Octanuclear cubic coordination cages. J. Am. Chem. Soc. 130, 15167–15175 (2008)
Stephenson, A., Sykes, D., Ward, M.D.: Cu12 and Cd16 coordination cages and their Cu3 and Cd3 subcomponents, and the role of inter-ligand π-stacking in stabilising cage complexes. Dalton Trans. 42, 6756 (2013)
Yan, X., Cook, T.R., Wang, P., Huang, F., Stang, P.J.: Highly emissive platinum(II) metallacages. Nat. Chem. 7, 342–348 (2015)
Frischmann, P.D., Kunz, V., Würther, F.: Bright fluorescence and host–guest sensing with a nanoscale M4L6 tetrahedron accessed by self-assembly of zinc–imine chelate vertices and perylene bisimide edges. Angew. Chem. Int. Ed. 54, 7285–7289 (2015)
Neelakandan, P.P., Jiménez, A., Nitschke, J.R.: Fluorophore incorporation allows nanomolar guest sensing and white-light emission in M4L6 cage complexes. Chem. Sci. 5, 908–915 (2014)
He, C., Lin, Z., Je, Z., Duan, C., Xu, C., Wang, Z., Yan, C.: A self-catalyzing hydrogen-storage material. Angew. Chem. Int. Ed. 47, 882–887 (2008)
Liu, Y., Wu, X., He, C., Li, Z., Duan, C.: Metal-organic polyhedra for selective sensing of ribonucleosides through the cooperation of hydrogen-bonding interactions. Dalton Trans. 39, 7727–7732 (2010)
Liu, Y., Wu, X., He, C., Zhang, R., Duan, C.: A truncated octahedral nanocage for fluorescent detection of nucleoside. Dalton Trans. 43, 5866–5868 (2008)
Noh, T.H., Hong, W., Lee, H., Jung, O.-S.: Indistinguishability and distinguishability between amide and ester moieties in the construction and properties of M6L8 octahedral nanocages. Dalton Trans. 44, 787–794 (2015)
Ghosh, S., Gole, B., Bar, A.K., Mukherjee, P.S.: Self-assembly of molecular prisms via Pt3 organometallic acceptors and a Pt2 organometallic clip. Organometallics. 28, 4288–4296 (2009)
Shanmugaraju, S., Jadhav, H., Patil, Y.P., Mukherjee, P.S.: Self-assembly of an octanuclear platinum(II) tetragonal prism from a new PtII4 organometallic star-shaped acceptor and its nitroaromatic sensing study. Inorg. Chem. 51, 13072–13074 (2012)
Wang, M., Vajpayee, V., Shanmugaraju, S., Zhen, Y.-R., Zhao, Z., Kim, H., Mukherjee, P.S., Chi, K.-W., Stang, P.J.: Coordination-driven self-assembly of M3L2 trigonal cages from preorganized metalloligands incorporating octahedral metal centers and fluorescent detection of nitroaromatics. Inorg. Chem. 50, 1506–1512 (2011)
Shanmugaraju, S., Mukherjee, P.S.: Self-assembled discrete molecules for sensing nitroaromatics. Chem. Eur. J. 21, 6656 (2015)
Han, M., Engelhard, D.M., Clever, G.H.: Self-assembled coordination cages based on banana-shaped ligands. Chem. Soc. Rev. 43, 1848 (2014)
Johnson, A.M., Moshe, O., Gamboa, A.S., Langloss, B.W., Limtiaco, J.F.K., Larive, C.K., Hooley, R.J.: Synthesis and properties of metal–ligand complexes with endohedral amine functionality. Inorg. Chem. 50, 9430–9442 (2011)
Kishi, N., Li, Z., Yoza, K., Akita, M., Yoshizawa, M.: An M2L4 molecular capsule with an anthracene Shell: encapsulation of large guests up to 1 nm. J. Am. Chem. Soc. 133, 11438–11441 (2011)
Kishi, N., Li, Z., Sei, Y., Akita, M., Yoza, K., Siegel, J.S., Yoshizawa, M.: Wide-ranging host capability of a PdII-linked M2L4 molecular capsule with an anthracene shell. Chem. Eur. J. 19, 6313–6320 (2013)
Li, Z., Kishi, N., Hasegawa, K., Akita, M., Yoshizawa, M.: Highly fluorescent M2L4 molecular capsules with anthracene shells. Chem. Commun. 47, 8605–8607 (2011)
Li, Z., Kishi, N., Yoza, K., Akita, M., Yoshizawa, M.: Isostructural M2L4 molecular capsules with anthracene shells: synthesis, crystal structures, and fluorescent properties. Chem. Eur. J. 18, 8358–8365 (2012)
Kishi, N., Akita, M., Yoshizawa, M.: Selective host–guest interactions of a transformable coordination capsule/tube with fullerenes. Angew. Chem. Int. Ed. 53, 3604 (2014)
Lewis, J.E.M., Elliott, A.B..S., McAdam, C.J., Gordon, K.C., Crowley, J.D.: ‘Click’ to functionalise: synthesis, characterisation and enhancement of the physical properties of a series of exo- and endo-functionalised Pd2L4nanocages. Chem. Sci. 5, 1833–1843 (2014)
Wang, C., Hao, X.-Q., Wang, M., Guo, C., Xu, B., Tan, E.N., Zhang, Y.-Y., Yu, Y., Li, Z.-Y., Yang, H.-B., Song, M.-P., Li, X.: Self-assembly of giant supramolecular cubes with terpyridine ligands as vertices and metals on edges. Chem. Sci. 5, 1221–1226 (2014)
Harano, K., Hiraoka, S., Shionoya, M.: Triangular and tetrahedral array of silver(I) ions by a novel disk-shaped tridentate ligand: dynamic control of coordination equilibrium of the silver(I) complexes. J. Am. Chem. Soc. 129, 5300–5301 (2007)
Frischmann, P.D., Mahata, K., Würthner, F.: Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem. Soc. Rev. 42, 1847–1870 (2013)
Wasielewski, M.R.: Self-assembly strategies for integrating light harvesting and charge separation in artificial photosynthetic systems. Acc. Chem. Res. 42, 1910–1921 (2009)
Durot, S., Taesch, J., Heitz, V.: Multiporphyrinic cages: architectures and functions. Chem. Rev. 114, 8542–8578 (2014)
Murase, T., Sato, S., Fujita, M.: Switching the interior hydrophobicity of a self-assembled spherical complex through the photoisomerization of confined azobenzene chromophores. Angew. Chem. Int. Ed. 46, 5133–5136 (2007)
Han, M., Michel, R., He, B., Chen, Y.-S., Stalke, D., John, M., Clever, G.H.: Light-triggered guest uptake and release by a photochromic coordination cage. Angew. Chem. Int. Ed. 52, 1319–1323 (2013)
Samanta, D., Mukherjee, P.S.: Sunlight-induced covalent marriage of two triply interlocked Pd6 cages and their facile thermal separation. J. Am. Chem. Soc. 136, 17006–17009 (2014)
Schmitt, F., Freudenreich, J., Barry, N.P.E., Juillerat-Jeanneret, L., Süss-Fink, G., Therrien, B.: Organometallic cages as vehicles for intracellular release of photosensitizers. J. Am. Chem. Soc. 134, 754–757 (2012)
Therrien, B.: Transporting and shielding Photosensitisers by using water-soluble organometallic cages: a new strategy in drug delivery and photodynamic therapy. Chem. Eur. J. 19, 8378–8386 (2013)
Roy, B., Ghosh, A.K., Srivastava, S., D’Silva, P., Mukherjee, P.S.: A Pd8 tetrafacial molecular barrel as carrier for water insoluble fluorophore. J. Am. Chem. Soc. 137, 11916 (2015)
Yamashina, M., Sartin, M., Sei, Y., Akita, M., Takeuchi, S., Tahara, T., Yoshizawa, M.: Preparation of highly fluorescent host–guest complexes with tunable color upon encapsulation. J. Am. Chem. Soc. 137, 9266 (2015)
Takezawa, H., Akiba, S., Murase, T., Fujita, M.: Cavity-directed chromism of phthalein dyes. J. Am. Chem. Soc. 137, 7043–7046 (2015)
Turega, S., Cullen, W., Whitehead, M., Hunter, C.A., Ward, M.D.: Mapping the internal recognition surface of an octanuclear coordination cage using guest libraries. J. Am. Chem. Soc. 136, 8475–8483 (2014)
Yang, Y., Chen, J.-S., Liu, J.-Y., Zhao, G.-J., Liu, L., Han, K.-L., Cook, T.R., Stang, P.: Photophysical properties of a post-self-assembly host/guest coordination cage: visible light driven Core-to-cage charge transfer. J. Phys. Chem. Lett. 6, 1942–1947 (2015)
Nishioka, Y., Yamaguchi, T., Yoshizawa, M., Fujita, M.: Unusual [2+4] and [2+2] cycloadditions of arenes in the confined cavity of self-assembled cages. J. Am. Chem. Soc. 129, 7000–7001 (2007)
Yoshizawa, M., Takeyama, Y., Okano, T., Fujita, M.: Cavity-directed synthesis within a self-assembled coordination cage: highly selective [2 + 2] cross-photodimerization of olefins. J. Am. Chem. Soc. 125, 3243–3247 (2003)
Nishioka, Y., Yamaguchi, T., Kawano, M., Fujita, M.: Asymmetric [2 + 2] olefin cross photoaddition in a self-assembled host with remote chiral auxiliaries. J. Am. Chem. Soc. 130, 8160–8161 (2008)
Furusawa, T., Kawano, M., Fujita, M.: The confined cavity of a coordination cage suppresses the photocleavage of alpha-diketones to give cyclization products through kinetically unfavorable pathways. Angew. Chem. Int. Ed. 46, 5717–5719 (2007)
Yamaguchi, T., Fujita, M.: Highly selective photomediated 1,4-radical addition to o-quinones controlled by a self-assembled cage. Angew. Chem. Int. Ed. 47, 2067–2069 (2008)
Karthikeyan, S., Ramamurthy, V.: Self-assembled coordination cage as a reaction vessel: triplet sensitized [2+2] photodimerization of acenaphthylene, and [4+4] photodimerization of 9-anthraldehyde. Tetrahedron Lett. 46, 4495–4498 (2005)
Karthikeyan, S., Ramamurthy, V.: Templating photodimerization of coumarins within a water-soluble nano reaction vessel. J. Org. Chem. 71, 6409–6413 (2006)
Karthikeyan, S., Ramamurthy, V.: Templating photodimerization of trans-cinnamic acid esters with a water-soluble Pd nanocage. J. Org. Chem. 72, 452–458 (2007)
Yoshizawa, M., Miyagi, S., Kawano, M., Ishiguro, K., Fujita, M.C.: Alkane oxidation via photochemical excitation of a self-assembled molecular cage. J. Am. Chem. Soc. 126, 9172–9173 (2004)
Furutani, Y., Kandori, H., Kawano, M., Nakabayashi, K., Yoshizawa, M., Fujita, M.: In situ spectroscopic, electrochemical, and theoretical studies of the photoinduced host−guest electron transfer that precedes unusual host-mediated alkane photooxidation. J. Am. Chem. Soc. 131, 4764–4768 (2009)
Murase, T., Nishijima, Y., Fujita, M.: Cytotoxicity of methanol extracts of Elaeis guineensis on MCF-7 and Vero cell lines. Chem. Asian J. 2, 826–829 (2012)
Murase, T., Takezawa, H., Fujita, M.: Photo-driven anti-Markovnikov alkyne hydration in self-assembled hollow complexes. Chem. Commun. 47, 10960–10962 (2011)
Klosterman, J.K., Iwamura, M., Tahara, T., Fujita, M.: Energy transfer in a mechanically trapped exciplex. J. Am. Chem. Soc. 131, 9478–9479 (2009)
Gera, R., Das, A., Jha, A., Dasgupta, J.: Light-induced proton-coupled electron transfer inside a nanocage. J. Am. Chem. Soc. 136, 15909–15912 (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nastasi, F., La Ganga, G., Di Pietro, M.L., Serroni, S., Campagna, S., Puntoriero, F. (2022). Multinuclear Metal Complexes: Coordination Dendrimers, Polymers, and Coordination Cages. In: Bahnemann, D., Patrocinio, A.O.T. (eds) Springer Handbook of Inorganic Photochemistry. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-030-63713-2_24
Download citation
DOI: https://doi.org/10.1007/978-3-030-63713-2_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63712-5
Online ISBN: 978-3-030-63713-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)