
Abstract
The most recent updated classification of inborn errors of immunity/primary immunodeficiencies, compiled by the International Union of Immunological Societies Expert Committee, was published in January 2020. Within days of completing this report, it was already out of date, evidenced by the frequent publication of genetic variants proposed to cause novel inborn errors of immunity. As the next formal report from the IUIS Expert Committee will not be published until 2022, we felt it important to provide the community with a brief update of recent contributions to the field of inborn errors of immunity. Herein, we highlight studies that have identified 26 additional monogenic gene defects that reach the threshold to represent novel causes of immune defects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
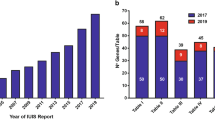
Inborn errors of immunity (IEI) are generally considered to result from monogenic germline defects that manifest as increased susceptibility to severe and/or recurrent infectious diseases, autoimmune or autoinflammatory conditions, atopic manifestations, and hematopoietic or solid tissue malignancies [1]. Over the past decade, the discovery of new IEIs has been occurring at an impressive rate. Indeed, the 2011 biennial update published by the IUIS Committee update listed 191 IEIs; this number increased to 430 in the 2019 update [2, 3]. This near-exponential increase in gene discovery is being driven by the accessibility and affordability of next-generation sequencing, and the efficient application of these technologies to elucidate the molecular etiology of unsolved cases of IEIs that are likely to result from single-gene defects [4].
Over the last 12 months, we have witnessed the ongoing rapid identification, and occasionally detailed molecular, biochemical, and cellular characterization, of genetic variants that cause, or are at least associated with, human diseases impacting host defense or immune regulation. Here, we will summarize reports on variants detected in 26 genes which we consider represent novel IEI (Table 1). Many additional genetic variants have been reported recently. However, those listed here have been adjudicated by the IUIS Committee to meet the strict criteria to be considered disease-causing [57]. These criteria include:
-
1.
The patient’s candidate genotype is monogenic and must not occur in individuals without the clinical phenotype;
-
2.
Experimental studies must indicate the genetic variant impairs, destroys, or alters expression or function of the gene product;
-
3.
The causal relationship between the candidate genotype and the clinical phenotype must be confirmed via a relevant cellular phenotype, including—where possible—rescue of a functional defect by reconstitution with the wild-type gene, or via a relevant animal phenotype [57].
We also considered (i) the numbers of individuals affected by the novel variants, (ii) sufficient justification for excluding alternative candidate gene variants identified in single cases especially in situations of consanguinity with recessive disease, (iii) the depth of the clinical descriptions of affected individuals, and (iv) the level of immune and mechanistic characterization.
Novel Causes of Inborn Errors of Immunity
Currently, inborn errors of immunity are listed in 10 tables: Immunodeficiencies affecting cellular and humoral immunity (Table I), Combined immunodeficiencies (CID) with syndromic features (Table II), Predominantly antibody deficiencies (Table III), Diseases of immune dysregulation (Table IV), Congenital defects of phagocytes (Table V), Defects in intrinsic and innate immunity (Table VI), Autoinflammatory diseases (Table VII), Complement deficiencies (Table VIII), Bone Marrow failure (Table IX), and Phenocopies of inborn errors of immunity (Table X). Several of these tables are further partitioned into various subtables (e.g., Table I is split into Subtable 1 [T−B+ Severe Combined Immune Deficiency (SCID)], Subtable 2 [T−B− SCID] and Subtable 3 [CID, generally less profound than SCID]) [2, 3].
Recently-reported gene defects have been found for most categories of inborn errors of immunity, including novel causes of:
-
Predominantly antibody deficiencies (FNIP1 [14, 15], PIK3CG [16, 17], CTNNBL1 [18], TNFSF13 [19]);
-
Autoinflammatory diseases (SOCS1 [20,21,22], TET2 [23], CEBPE [24], CDC42 [33,34,35,36,37,38,39], LSM11, RNU7–1 [32], STAT2 [40, 41], RIPK1 [42, 43], NCKAP1L [44,45,46]), UBA1 (somatic mutations) [47]; and
-
Susceptibility to infection with specific pathogens (MAPK8 [31]; TBX21 [25], IFNG [26], NOS2 [28], SNORA31 [29], ATG4A, MAP1LC3B2 [30]) (Table 1).
Notably, several of these genes are already included in previous IUIS updates, namely IL6ST, STAT2, CEBPE, and RIPK1 [2, 3]. However, they are listed here because the variant identified is pathogenic via a distinct mechanism and/or different mode of inheritance; i.e., autosomal recessive (AR) vs autosomal dominant for IL6ST [9] or RIPK1 [42, 43], partial deficiency vs complete deficiency for IL6ST [10, 11], or AR loss of function vs AR gain of function for CEBPE [24] or STAT2 [40, 41]. Furthermore, the GOF variants reported for CEBPE appear to represent the first described germline neomorphic mutation in inborn errors of immunity where the variant allele has completely novel functions not seen for the wild type gene [24]. Thus, these findings underscore the importance of appropriately interpreting genetic variants identified by next-generation sequencing, not discarding variants of unknown significance simply because they do not match the expected zygosity or clinical phenotype of previously reported studies, and to rigorously validate the impact of novel variants on the function of the encoded protein.
Joining the Dots with Discoveries of Novel Inborn Errors of Immunity
Many known inborn errors of immunity impact a defined signaling pathway such that mutations in components of these same pathways can represent clinical phenocopies of diseases causes by distinct genetic variants (genetic heterogeneity). In other words, physiological homogeneity can be identified for many genotypes underlying a given phenotype. Classic examples of this are Mendelian susceptibility to mycobacterial disease (MSMD), which results from impaired IFNγ-mediated immunity following exposure to mycobacterial species [58], and herpes simplex virus encephalitis (HSE) resulting from impaired TLR3-mediated anti-HSV1 immunity [59, 60]. Thus, variants in genes affecting the production of IFNγ (e.g., IL12RB1, IL12RB2, IL23R, TYK2, IKBKG, SPPL2A, IRF8) or cellular responses to IFNγ (e.g., IFNGR1, IFNGR2, STAT1, JAK1) result in MSMD in otherwise healthy individuals [58]. Similarly, inactivating mutations in signaling components of the TLR3 signaling pathway (TLR3, UNC93B, TRIF, TRAF3, TBK1, IRF3) underlie HSE due to impaired type 1 IFN-mediated central nervous system (CNS) intrinsic immunity against HSV1 [59, 60].
Recent discoveries have further linked common clinical phenotypes with unique genotypes that converge in a shared pathway. Thus, the non-redundant role of IFNγ-mediated immunity in host defense against mycobacterial infection [58] has been definitively established by the identification of individuals with inactivating bi-allelic mutations in not only IFNG itself [26] but also TBX21 [25], the transcription factor that regulates expression and production of IFNγ.
Interestingly, variants in the small nucleolar RNA SNORA31 predispose affected individuals to HSE. Mechanistically, patient’s iPSC-derived cortical neurons were found to be highly susceptible to HSV-1 infection in vitro, and this could be restored by exogenous IFNβ [29, 60]. However, responses of these cells to TLR3 and IFNβ, but not HSV1, are intact, revealing that SNORA31 functions to regulate cell-intrinsic immunity to HSV-1 by a mechanism independent of TLR3 signaling [29, 60]. The discovery of individuals with SNORA31 variants will facilitate further understanding of CNS-intrinsic host defense.
The discoveries of individuals with complete gp130-deficiency due to null/nonsense bi-allelic mutations of IL6ST [11], or pathogenic dominant-negative heterozygous variants of IL6ST [9], and a phenotype of eczema, hyper-IgE, and eosinophilia, likely explain these features of autosomal dominant hyper-IgE syndrome due to STAT3 negative dominance [61] and further highlight the role of IL-6 signaling in restraining atopic and allergic responses. Furthermore, the lack of mucocutaneous candidiasis in patients with impaired signaling via receptors for IL-6 (IL6R, IL6ST mutations [9, 11, 50, 62, 63]; anti-IL-6 autoantibodies [64]), IL-23 (biallelic IL23R variants) [65] or IL-21 (biallelic IL21 or IL21R variants) [66] argues that individually these cytokines are not required for the STAT3-mediated generation of human Th17 cells and host defense against fungal infections. Rather, the combinatorial defect of impaired STAT3 signaling downstream of these receptors explains chronic mucocutaneous candidiasis in an individual with dominant-negative STAT3 mutations. These findings again reveal the capacity for inborn errors of immunity to provide convincing evidence for basic immunological concepts. Indeed, this is further exemplified by the discovery that variants of ATG4A or MAP1LC3B2 cause recurrent HSV2 infection of the CNS, thereby establishing hitherto non-redundant functions of the autophagy pathway in non-hematopoietic cell-mediated intrinsic anti-viral immune responses [30].
SARS-CoV2 and Inborn Errors of Immunity
The COVID19 pandemic of 2020 has clearly changed the world in many ways. It has also yielded opportunities to understand host requirements for immunity against SARS-CoV2 infection. A recent study of ~ 650 individuals who developed severe COVID-19 found that ~ 3.5% of patients harbored germline loss-of-function variants in genes previously found to be important for host defense against influenza or other viral infections (e.g., bi-allelic loss of function mutations of IRF7 or IFNAR1, heterozygous mutations in TLR3, TICAM1, TBK1, or IRF3) [67] due to the key role of these genes in the type 1 IFN signaling pathway [59, 68]. An accompanying study found that, strikingly, ~ 10% of patients with severe COVID-19 have high levels of neutralizing autoantibodies (autoAbs) against type 1 IFNs in their serum [48]. The impact of these autoAbs was evidenced by the inability to detect IFN in serum from these patients, and their capacity to prevent anti-viral immune responses in vitro [48] (Table 1). These studies defined a crucial and non-redundant role for type 1 IFNs in immune control of SARS-CoV2 infection, and thus prevention of severe COVID-19. Furthermore, they also established that autoAbs against type 1 IFN phenocopy an inborn error of immunity, as previously determined for autoAbs against IFNγ and susceptibility to mycobacterial disease, anti-Th17 cytokine (IL-17A, IL-17F, IL-22) autoAbs in individuals with chronic mucocutaneous candidiasis, or pyogenic infections due to anti-IL-6 autoAbs [64, 69].
Conclusions
Discoveries over the past 12 months in the field of inborn errors of immunity have further identified non-redundant functions of key genes in human immune cell development, host defense, and immune regulation. In some cases, these functions go well beyond what may have been expected or anticipated based on animal models (e.g., TBX21 [25]). They have also already highlighted the heterogenous phenotypes that can result from variants in the same gene (e.g., CDC42 [33,34,35,36,37,38,39, 52]), indicated that significant diseases can arise from mono-allelic or bi-allelic loss of function (IL6ST [9], RIPK1 [42, 43]) or bi-allelic loss- or gain-of-function (CEBPE [24], STAT2 [40, 41]) variants in the same gene, or from autoAb phenocopies of monogenic lesions (e.g., COVID19 and anti-IFN Abs) [48], and identified novel somatic mutations as pathogenic causes of immune disorders (UBA1) [47]. Importantly, they have also provided opportunities for therapeutic interventions, such as JAK inhibitors to treat STAT2 gain of function [40, 41] or SOCS1 deficiency [22], IFNγ to treat mycobacterial disease [25, 26], or early IFN-β or IFN-α2a treatment of SARS-CoV2 infection in COVID-19 patients with autoantibodies against IFN-α or IFN-ω [67] or impaired type 1 IFN responses [70]. This snapshot of genetic discoveries underpinning human immune disorders further highlights the critical contributions of inborn errors of immunity to our broader understanding of basic, translational, and clinical immunology.
Data Availability
Not applicable.
References
Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human inborn errors of immunity: an expanding universe. Sci Immunol. 2020;5(49):eabb1662. https://doi.org/10.1126/sciimmunol.abb1662.
Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;40(1):66–81. https://doi.org/10.1007/s10875-020-00758-x.
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. https://doi.org/10.1007/s10875-019-00737-x.
Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016;138(4):957–69. https://doi.org/10.1016/j.jaci.2016.08.003.
Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. 2020;5(44):eaax1036. https://doi.org/10.1126/sciimmunol.aax1036.
Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. 2017;92(6):664–8. https://doi.org/10.1111/cge.13085.
Lev A, Lee YN, Sun G, Hallumi E, Simon AJ, Zrihen KS, et al. Inherited SLP76 deficiency in humans causes severe combined immunodeficiency, neutrophil and platelet defects. J Exp Med. 2021;218(3):e20201062. https://doi.org/10.1084/jem.20201062.
Mace EM, Paust S, Conte MI, Baxley RM, Schmit MM, Patil SL, et al. Human NK cell deficiency as a result of biallelic mutations in MCM10. J Clin Invest. 2020;130:5272–86. https://doi.org/10.1172/JCI134966.
Beziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. 2020;217(6):e20191804. https://doi.org/10.1084/jem.20191804.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104(6):1182–201. https://doi.org/10.1016/j.ajhg.2019.04.011.
Chen YH, Grigelioniene G, Newton PT, Gullander J, Elfving M, Hammarsjo A, et al. Absence of GP130 cytokine receptor signaling causes extended Stuve-Wiedemann syndrome. J Exp Med. 2020;217(3):e20191306. https://doi.org/10.1084/jem.20191306.
Park H, Staehling K, Tsang M, Appleby MW, Brunkow ME, Margineantu D, et al. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity. 2012;36(5):769–81. https://doi.org/10.1016/j.immuni.2012.02.019.
Siggs OM, Stockenhuber A, Deobagkar-Lele M, Bull KR, Crockford TL, Kingston BL, et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc Natl Acad Sci U S A. 2016;113(26):E3706–15. https://doi.org/10.1073/pnas.1607592113.
Niehues T, Ozgur TT, Bickes M, Waldmann R, Schoning J, Brasen J, et al. Mutations of the gene FNIP1 associated with a syndromic autosomal recessive immunodeficiency with cardiomyopathy and pre-excitation syndrome. Eur J Immunol. 2020;50(7):1078–80. https://doi.org/10.1002/eji.201948504.
Saettini F, Poli C, Vengoechea J, Bonanomi S, Orellana JC, Fazio G, et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin interacting protein 1 deficiency. Blood. 2021;137:493–9 https://doi.org/10.1182/blood.2020006441.
Takeda AJ, Maher TJ, Zhang Y, Lanahan SM, Bucklin ML, Compton SR, et al. Human PI3Kgamma deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat Commun. 2019;10(1):4364. https://doi.org/10.1038/s41467-019-12311-5.
Thian M, Hoeger B, Kamnev A, Poyer F, Kostel Bal S, Caldera M, et al. Germline biallelic PIK3CG mutations in a multifaceted immunodeficiency with immune dysregulation. Haematologica. 2020;105:e488. https://doi.org/10.3324/haematol.2019.231399.
Kuhny M, Forbes LR, Cakan E, Vega-Loza A, Kostiuk V, Dinesh RK, et al. Disease-associated CTNNBL1 mutation impairs somatic hypermutation by decreasing nuclear AID. J Clin Invest. 2020;130(8):4411-4422. https://doi.org/10.1172/JCI131297.
Yeh TW, Okano T, Naruto T, Yamashita M, Okamura M, Tanita K, et al. APRIL-dependent life-long plasmacyte maintenance and immunoglobulin production in humans. J Allergy Clin Immunol. 2020;146:1109–1120.e4. https://doi.org/10.1016/j.jaci.2020.03.025.
Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. 2020;146(5):1194–1200.e1. https://doi.org/10.1016/j.jaci.2020.07.033.
Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020;583(7814):90–5. https://doi.org/10.1038/s41586-020-2265-1.
Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. 2020;11(1):5341. https://doi.org/10.1038/s41467-020-18925-4.
Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood. 2020;136(9):1055–66. https://doi.org/10.1182/blood.2020005844.
Goos H, Fogarty CL, Sahu B, Plagnol V, Rajamaki K, Nurmi K, et al. Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J Allergy Clin Immunol. 2019;144(5):1364–76. https://doi.org/10.1016/j.jaci.2019.06.003.
Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, et al. Human T-bet governs innate and innate-like adaptive IFN-gamma immunity against mycobacteria. Cell. 2020;183(7):1826–1847.e31. https://doi.org/10.1016/j.cell.2020.10.046.
Kerner G, Rosain J, Guerin A, AlKhabaz A, Oleaga-Quintas C, Rapaport F, et al. Inherited human IFNgamma deficiency underlies mycobacterial disease. J Clin Invest. 2020;130(6):3158–71. https://doi.org/10.1172/JCI135460.
Noda S, Tanaka K, Sawamura S, Sasaki M, Matsumoto T, Mikami K, et al. Role of nitric oxide synthase type 2 in acute infection with murine cytomegalovirus. J Immunol. 2001;166(5):3533–41. https://doi.org/10.4049/jimmunol.166.5.3533.
Drutman SB, Mansouri D, Mahdaviani SA, Neehus AL, Hum D, Bryk R, et al. Fatal Cytomegalovirus infection in an adult with inherited NOS2 deficiency. N Engl J Med. 2020;382(5):437–45. https://doi.org/10.1056/NEJMoa1910640.
Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, et al. Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat Med. 2019;25(12):1873–84. https://doi.org/10.1038/s41591-019-0672-3.
Hait AS, Olagnier D, Sancho-Shimizu V, Skipper KA, Helleberg M, Larsen SM, et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci Immunol. 2020;5(54):eabc2691. https://doi.org/10.1126/sciimmunol.abc2691.
Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci Immunol. 2019;4(41):eabc2691. https://doi.org/10.1126/sciimmunol.aax7965.
Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. 2020;52(12):1364–72. https://doi.org/10.1038/s41588-020-00737-3.
Verboon JM, Mahmut D, Kim AR, Nakamura M, Abdulhay NJ, Nandakumar SK, et al. Infantile myelofibrosis and myeloproliferation with CDC42 dysfunction. J Clin Immunol. 2020;40:554–66. https://doi.org/10.1007/s10875-020-00778-7.
Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. 2019;216(12):2778–99. https://doi.org/10.1084/jem.20190147.
Gernez Y, de Jesus AA, Alsaleem H, Macaubas C, Roy A, Lovell D, et al. Severe autoinflammation in 4 patients with C-terminal variants in cell division control protein 42 homolog (CDC42) successfully treated with IL-1beta inhibition. J Allergy Clin Immunol. 2019;144(4):1122–5 e6. https://doi.org/10.1016/j.jaci.2019.06.017.
Bucciol G, Pillay B, Casas-Martin J, Delafontaine S, Proesmans M, Lorent N, et al. Systemic inflammation and myelofibrosis in a patient with Takenouchi-Kosaki syndrome due to CDC42 Tyr64Cys mutation. J Clin Immunol. 2020;40:567–70. https://doi.org/10.1007/s10875-020-00742-5.
Bekhouche B, Tourville A, Ravichandran Y, Tacine R, Abrami L, Dussiot M, et al. A toxic palmitoylation of Cdc42 enhances NF-kappaB signaling and drives a severe autoinflammatory syndrome. J Allergy Clin Immunol. 2020;146(5):1201–1204.e8. https://doi.org/10.1016/j.jaci.2020.03.020.
He T, Huang Y, Ling J, Yang J. A new patient with NOCARH syndrome due to CDC42 defect. J Clin Immunol. 2020;40(4):571–5. https://doi.org/10.1007/s10875-020-00786-7.
Szczawinska-Poplonyk A, Ploski R, Bernatowska E, Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: a case report. Front Immunol. 2020;11:318. https://doi.org/10.3389/fimmu.2020.00318.
Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med. 2020;217(5):e20192319. https://doi.org/10.1084/jem.20192319.
Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol. 2019;4(42):eaav7501. https://doi.org/10.1126/sciimmunol.aav7501.
Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577(7788):109–14. https://doi.org/10.1038/s41586-019-1830-y.
Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577(7788):103–8. https://doi.org/10.1038/s41586-019-1828-5.
Cook SA, Comrie WA, Poli MC, Similuk M, Oler AJ, Faruqi AJ, et al. HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease. Science. 2020;369(6500):202–7. https://doi.org/10.1126/science.aay5663.
Salzer E, Zoghi S, Kiss MG, Kage F, Rashkova C, Stahnke S, et al. The cytoskeletal regulator HEM1 governs B cell development and prevents autoimmunity. Sci Immunol. 2020;5(49):eabc3979. https://doi.org/10.1126/sciimmunol.abc3979.
Castro CN, Rosenzwajg M, Carapito R, Shahrooei M, Konantz M, Khan A, et al. NCKAP1L defects lead to a novel syndrome combining immunodeficiency, lymphoproliferation, and hyperinflammation. J Exp Med. 2020;217(12):e20192275. https://doi.org/10.1084/jem.20192275.
Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383:2628–38. https://doi.org/10.1056/NEJMoa2026834.
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4585. https://doi.org/10.1126/science.abd4585.
Pohl E, Aykut A, Beleggia F, Karaca E, Durmaz B, Keupp K, et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet. 2013;132(11):1311–20. https://doi.org/10.1007/s00439-013-1337-9.
Spencer S, Kostel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. 2019;216(9):1986–98. https://doi.org/10.1084/jem.20190344.
Gombart AF, Koeffler HP. Neutrophil specific granule deficiency and mutations in the gene encoding transcription factor C/EBP(epsilon). Curr Opin Hematol. 2002;9(1):36–42. https://doi.org/10.1097/00062752-200201000-00007.
Su HC, Orange JS. The growing spectrum of human diseases caused by inherited CDC42 mutations. J Clin Immunol. 2020;40(4):551–3. https://doi.org/10.1007/s10875-020-00785-8.
Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci U S A. 2013;110(8):3053–8. https://doi.org/10.1073/pnas.1220098110.
Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol. 2017;139(6):1995–7 e9. https://doi.org/10.1016/j.jaci.2016.10.033.
Cuchet-Lourenco D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science. 2018;361(6404):810–3. https://doi.org/10.1126/science.aar2641.
Li Y, Fuhrer M, Bahrami E, Socha P, Klaudel-Dreszler M, Bouzidi A, et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2019;116(3):970–5. https://doi.org/10.1073/pnas.1813582116.
Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211(11):2137–49. https://doi.org/10.1084/jem.20140520.
Bustamante J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. 2020;139(6–7):993–1000. https://doi.org/10.1007/s00439-020-02120-y.
Moens L, Meyts I. Recent human genetic errors of innate immunity leading to increased susceptibility to infection. Curr Opin Immunol. 2020;62:79–90. https://doi.org/10.1016/j.coi.2019.12.002.
Zhang SY. Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Hum Genet. 2020;139(6–7):911–8. https://doi.org/10.1007/s00439-020-02127-5.
Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–28. https://doi.org/10.1016/j.immuni.2012.03.016.
Shahin T, Aschenbrenner D, Cagdas D, Bal SK, Conde CD, Garncarz W, et al. Selective loss of function variants in IL6ST cause hyper-IgE syndrome with distinct impairments of T-cell phenotype and function. Haematologica. 2019;104(3):609–21. https://doi.org/10.3324/haematol.2018.194233.
Nahum A, Sharfe N, Broides A, Dadi H, Naghdi Z, Mandola AB, et al. Defining the biological responses of IL-6 by the study of a novel IL-6 receptor chain immunodeficiency. J Allergy Clin Immunol. 2020;145(3):1011–5 e6. https://doi.org/10.1016/j.jaci.2019.11.015.
Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, et al. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J Immunol. 2008;180(1):647–54. https://doi.org/10.4049/jimmunol.180.1.647.
Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F, et al. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. 2018;3(30):eaau6759. https://doi.org/10.1126/sciimmunol.aau6759.
Kotlarz D, Zietara N, Milner JD, Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr. 2014;26(6):704–12. https://doi.org/10.1097/MOP.0000000000000160.
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515):eabd4570. https://doi.org/10.1126/science.abd4570.
Zhang Q. Human genetics of life-threatening influenza pneumonitis. Hum Genet. 2020;139(6–7):941–8. https://doi.org/10.1007/s00439-019-02108-3.
Ku CL, Chi CY, von Bernuth H, Doffinger R. Autoantibodies against cytokines: phenocopies of primary immunodeficiencies? Hum Genet. 2020;139(6–7):783–94. https://doi.org/10.1007/s00439-020-02180-0.
Levy R, Bastard P, Lanternier F, Lecuit M, Zhang SY, Casanova JL. IFN-alpha2a therapy in two patients with inborn errors of TLR3 and IRF3 infected with SARS-CoV-2. J Clin Immunol. 2021;41:26–7. https://doi.org/10.1007/s10875-020-00933-0.
Funding
The members of the Inborn Errors of Immunity committee would like to thank the International Union of Immunological Societies for funding, as well as CSL Behring, Baxalta, and Shire/Takeda for providing educational grants to enable us to compile this interim update to novel causes of immune diseases. This work was also supported in part by the Intramural Research Program of the NIAID, NIH. SGT is supported by an Investigator Grant (Level 3) awarded by the National Health and Medical Research Council of Australia. IM is a senior clinical investigator of FWO Vlaanderen (EBD-D8974-FKM).
Author information
Authors and Affiliations
Contributions
SGT wrote the drafts of the manuscript, prepared the table, and revised the original manuscripts for resubmission. All co-authors contributed to and edited drafts of the original and revised manuscripts and table, and approved the final submitted version.
Corresponding author
Ethics declarations
Ethics Approval
This work is a review of recently-reported genetic variants that represent novel inborn errors of immunity. No human research studies were performed in order to produce this review. Thus, no approvals by appropriate institutional review boards or human research ethics committees were required to undertake the preparation of this report.
Consent to Participate
Not applicable as this is a review of recently-reported genetic variants.
Consent for Publication
The authors consent to publish the content of this review. However, as noted above, as this is a review of recently-reported genetic variants that represent novel inborn errors of immunity, we did not require consent to publish from participants.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tangye, S.G., Al-Herz, W., Bousfiha, A. et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: an Interim Update by the IUIS Committee. J Clin Immunol 41, 666–679 (2021). https://doi.org/10.1007/s10875-021-00980-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-021-00980-1