
Abstract
Herpes simplex virus 1 (HSV-1) encephalitis (HSE) is the most common sporadic viral encephalitis in Western countries. Over the last 15 years, human genetic and immunological studies have provided proof-of-principle that childhood HSE can result from inborn errors of central nervous system (CNS)-specific, cell-intrinsic immunity to HSV-1. HSE-causing mutations of eight genes disrupt known (TLR3-dependent IFN-α/β immunity) and novel (dependent on DBR1 or snoRNA31) antiviral mechanisms. Monogenic inborn errors confer susceptibility to forebrain (TLR3-IFN or snoRNA31) or brainstem (DBR1) HSE. Most of these disorders display incomplete clinical penetrance, with the possible exception of DBR1 deficiency. They account for a small, but non-negligible proportion of cases (about 7%). These findings pave the way for the gradual definition of the genetic and immunological architecture of childhood HSE, with both biological and clinical implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Herpes simplex virus 1 (HSV-1) encephalitis (HSE) is the most common sporadic viral encephalitis in Western countries and, perhaps, worldwide. The causal role of HSV in HSE was established in 1941 (Smith et al. 1941). HSE is considered the most severe clinical form of infection with HSV-1, an almost ubiquitous, and typically innocuous virus. HSV-1 is a double-stranded enveloped DNA virus. It enters the body via the oral or nasal epithelium and infects neurons, subsequently establishing latency in the trigeminal (TG) sensory ganglia (Smith 2012). More than 85% of adults worldwide have antibodies against HSV-1, which causes asymptomatic infection or benign, self-healing disease in most individuals, with gingivitis and stomatitis during primary infection and herpes labialis during reactivation (Whitley 2002; Whitley and Gnann 1993). In about 1 ~ 2/10,000 infected individuals of all ages, HSV-1 invades the central nervous system (CNS) via the olfactory bulb, causing forebrain HSE (~ 95% of cases) or, more rarely, via the TG nerves, causing brainstem HSE (~ 5% of cases) (Abel et al. 2010; Jubelt et al. 2011; Whitley 2006, 2015). In general, people suffer from HSE are otherwise healthy individuals, who are not particularly susceptible to other clinical forms of HSV-1 infection. No viral dissemination to the bloodstream or tissues other than the brain has been reported during the course of HSE (Gnann and Whitley 2017; Whitley 2015). About one-third of HSE cases occur in children, with incidence peaking between the ages of 3 months and 6 years, earlier than would be expected if the risk were independent of age at primary infection with HSV-1 (Abel et al. 2010; Hjalmarsson et al. 2007; Kelly and Kroll 2004; Kohl 1988, 1998; Raschilas et al. 2002; Schlesinger et al. 1995; Whitley 2002). HSE is fatal in more than 70% of the patients if untreated, and most acyclovir-treated survivors present mild to severe neurological sequelae (Campbell et al. 1982; Stahl and Mailles 2019). Beyond identification of the causal virus, the pathogenesis of HSE has long remained unclear.
In the early 2000s, it was hypothesized that childhood HSE might result from single-gene inborn errors of immunity to HSV-1 in the CNS (Alcais et al. 2010; Casanova 2015a, b). HSV-1 causes HSE in only a small proportion of HSV-1-infected individuals, and no HSE epidemics are observed, strongly suggesting a key role for inherited or acquired host susceptibility to HSV-1 in the pathogenesis of this condition. Moreover, although HSE is almost always sporadic, as opposed to epidemic or familial, four multiplex kindreds with an interval of several years between HSE cases have been reported (Gazquez et al. 1996; Jackson et al. 2002; Koskiniemi et al. 1995; Lerner et al. 1983). Furthermore, a high frequency of consanguinity among the parents of children with HSE was reported in a retrospective French survey (12%) (Abel et al. 2010). These findings suggested that HSE may be genetic, at least in some children. Remarkably, HSE has not been reported in children with AIDS or conventional PIDs, such as lymphopenia, agammaglobulinemia, or neutropenia, suggesting that inborn errors of leukocytes are unlikely to be causal (Notarangelo et al. 2006; Ochs et al. 2007; Tangye et al. 2020). Moreover, very rare cases of “syndromic” HSE have been observed in combination with mycobacterial diseases, in a child with autosomal recessive (AR) STAT1 deficiency (Dupuis et al. 2003), and a child with a NEMO mutation (Audry et al. 2011; Niehues et al. 2004; Puel et al. 2006). Both disorders impair cell-intrinsic immunity in leukocytes and, probably, in all other cell types. The cells of the STAT-1-deficient patient displayed impaired responses to interferon (IFN) IFN-α/β, IFN-λ and IFN-γ (Chapgier et al. 2006, 2009; Dupuis et al. 2003), whereas the cells of the patient with the NEMO mutation displayed poor IFN-α/β and IFN-λ production (Audry et al. 2011). These observations suggested that human genetic defects affecting intrinsic immunity in the CNS, probably related to IFN-mediated antiviral defense, may underlie HSE, at least in some children. This led to the discovery of human genetic lesions underlying the typical form of isolated childhood HSE.
Inborn errors of TLR3 immunity and forebrain HSE
Toll-like receptor 3 (TLR3) is an endosomal receptor of dsRNA (Alexopoulou et al. 2001), and an inducer of IFN-α/β and -λ. It is highly conserved across vertebrate species (Mikami et al. 2012) and has evolved under strong purifying selection in humans (Barreiro et al. 2009). Upon ligand binding, TLR3 recruits a single adaptor, TRIF, inducing the production of antiviral cytokines, including IFN-β, IFN-λ, and inflammatory cytokines, such as TNF-α and IL-6, through the activation of transcription factors, including IRF3 and NF-κB (Kawai and Akira 2006). Genetic studies of isolated HSE of the forebrain led to the discovery of single-gene inborn errors of the TLR3-dependent pathway of IFN-α/β and -λ induction, with mono- or biallelic mutations of six TLR3 pathway genes (TLR3, UNC93B1, TRIF, TRAF3, TBK1 or IRF3) (Andersen et al. 2015; Casrouge et al. 2006; Guo et al. 2011; Herman et al. 2012; Lim et al. 2014; Perez de Diego et al. 2010; Sancho-Shimizu et al. 2011; Zhang and Casanova 2015; Zhang et al. 2007) (Table 1). These findings, together with the previous observation of syndromic HSE in patients with X-linked recessive (XR) NEMO deficiency (Audry et al. 2011) or AR complete STAT1 deficiency (Dupuis et al. 2003), suggested that TLR3-dependent IFN-α/β and/or -λ immunity is crucial for host defense against HSV-1 in the CNS. It has also been suggested that other mutations of these and other TLR3 or IFN pathway genes may underlie HSE in children or adults (Armangue et al. 2019; Garcia-Morato et al. 2019; Mork et al. 2015; Sironi et al. 2017; Vitturi et al. 2019). Interestingly, a few other patients with mutations in TLR3 developed influenza A virus pneumonitis or varicella zoster virus ophthalmicus (Lim et al. 2019; Liang et al. 2019). Nevertheless, TLR3-mediated responses to dsRNA and antiviral immunity seem to be redundant in most TLR3-expressing cell types, including leukocytes in particular, probably accounting for the lack of viral dissemination during the course of HSE (Guo et al. 2011; Zhang et al. 2007). The hypothesis that CNS-specific cell-intrinsic immunity rather than leukocyte-mediated immunity is crucial for host defense against HSV-1 was tested experimentally, initially with dermal fibroblasts as surrogate cells, and then with induced pluripotent stem cell (iPSC)-derived CNS- and peripheral nervous system (PNS)-resident cells from patients with forebrain HSE and mutations of TLR3 pathway genes. Like what was shown in recently studies that mouse Tlr3 is required for responses to HSV in neurons and astrocytes (Reinert et al. 2012; Sato et al. 2018), TLR3 pathway-deficient human fibroblasts (Casrouge et al. 2006; Guo et al. 2011; Herman et al. 2012; Lim et al. 2014; Perez de Diego et al. 2010; Sancho-Shimizu et al. 2011; Zhang et al. 2007) and iPSC-derived cortical neurons and oligodendrocytes (Lafaille et al. 2012) were found to be much more susceptible to HSV-1 infection than control cells. This phenotype was rescued by the addition of exogenous IFN-a/β but not IFN-l. By contrast, in vitro-differentiated human UNC-93B-deficient astrocytes or neural stem cells, and TLR3-deficient peripheral TG neurons had a susceptibility to infection similar to that of control cells (Zimmer et al. 2018). Microglial cells, the CNS-resident macrophages that have been shown to rely on the cGAS/STING pathway to orchestrate anti-HSV-1 defense in mice (Reinert et al. 2016), have not yet been tested in the setting of human immunology to HSV-1 infection. TLR3-dependent, IFN-mediated cortical neuron- and oligodendrocyte-autonomous anti-HSV-1 immunity thus appears to be crucial for host defense against HSV-1 infection in the human forebrain. These data provided a plausible cellular basis for the pathogenesis of genetically driven forebrain HSE, suggesting that cell-intrinsic immunity, as opposed to the innate and adaptive immunity mediated by leukocytes and related cells, was crucial for host defense against HSV-1 in the human forebrain.
Inborn errors of snoRNA31 and forebrain HSE
Small nucleolar RNA 31 (SnoRNA31) is a 130-nucleotide snoRNA of the H/ACA box class. The gene encoding this molecule, SNORA31, is highly conserved in the general population, and ubiquitously expressed in various human cell types (Jorjani et al. 2016). SnoRNA31 has only one predicted function: directing the isomerization of uridine residues to pseudouridine in position 218 of the 18S ribosomal RNA (rRNA) and position 3713 of the 28S rRNA (Kiss 2004). Five unrelated patients with forebrain HSE have each been shown to carry one of four rare heterozygous variants of SNORA31 (Lafaille et al. 2019) (Table 1). Studies with human pluripotent stem cell (hPSC)-derived cortical neurons showed that snoRNA31 is produced and functional in human cortical neurons, as a CRISPR/Cas9-introduced biallelic deletion in SNORA31 impairs pseudouridylation of the uridine residue in position 218 of the rRNA 18S in isogenic hPSC-derived CNS neurons. Moreover, snoRNA31 is a CNS neuron-intrinsic HSV-1 restriction factor, as CRISPR/Cas9-introduced biallelic and monoallelic SNORA31 deletions render neurons highly susceptible to HSV-1. Accordingly, CNS cortical neurons derived from the iPSCs of patients with SNORA31 mutations are highly susceptible to HSV-1, like those from TLR3- or STAT1-deficient patients. Exogenous IFN-β renders SNORA31- and TLR3-, but not STAT1-mutated neurons resistant to HSV-1 infection. Finally, transcriptomic analyses of SNORA31-mutated hPSC-derived cortical neurons have shown that these cells have normal responses to TLR3 and IFN-α/β stimulations, but abnormal responses to HSV-1, suggesting that AD snoRNA31 deficiency impairs intrinsic immunity by a distinctive mechanism in vitro, and by inference underlies HSE in vivo by a distinctive mechanism. The modified transcriptome level response to HSV-1 associated with snoRNA31 deficiency may affect the expression of one or more of the effectors induced by TLR3 or IFN-α/β, thereby impairing anti-HSV-1 immunity in these cells. Alternatively, snoRNA31 may interfere with HSV-1 propagation directly, by interacting with viral transcripts. Future studies should address the fine molecular mechanism by which snoRNA31 contributes to the control of HSV-1 in CNS neurons and, potentially, in other CNS-resident cell types. The discovery of AD snoRNA31 deficiency as a new genetic etiology of forebrain HSE demonstrated the essential role of human snoRNA31 in CNS neuron-intrinsic immunity to HSV-1, providing evidence that snoRNAs can be essential for host defense.
Inborn errors of DBR1 and brainstem HSE
Debranching enzyme 1 (DBR1) is the only known RNA lariat debranching enzyme in humans. As inferred from studies performed in yeast, DBR1 hydrolyzes 2′5′-phosphodiester linkages at the branch points of intron lariat RNAs, facilitating their rapid turnover (Arenas and Hurwitz 1987; Chapman and Boeke 1991; Jacquier and Rosbash 1986; Nam et al. 1994, 1997; Ruskin and Green 1985). No connection between DBR1 and host immunity to infection was known. In this context, AR partial DBR1 deficiency has recently been reported in otherwise healthy children with brainstem viral encephalitis (BVE) caused by various viruses, including HSV-1, influenza B virus (IBV), and norovirus (Zhang et al. 2018) (Table 1). In most patients with devastating BVE, the brainstem is the only region of the CNS affected, suggesting that, if there is an inborn error of immunity underlying BVE, it may affect brainstem-specific immunity. Two of the five DBR1-deficient children developed brainstem HSE. DBR1 protein levels are highest in the brainstem and spinal cord, suggesting that DBR1 deficiency disrupts immunity in brainstem-resident cells (Zhang et al. 2018). DBR1-deficient fibroblasts from the patients, whose TLR3 and IFN-α/β responsive pathways were intact, were found to contain higher RNA lariat levels than control cells, this difference becoming even more marked during HSV-1 infection. Moreover, DBR1-deficient fibroblasts were highly susceptible to HSV-1 and vesicular stomatitis virus, like TLR3- and STAT1-deficient fibroblasts (Zhang et al. 2018). The underlying molecular mechanism remains elusive. The accumulation of RNA lariats may impair virus recognition by host cells, thereby damaging cell-intrinsic defenses against viral invasion. DBR1 may also regulate the processing of some host protein-coding RNAs, non-coding RNAs (Han et al. 2017; Murray et al. 2014; Ooi et al. 1998; Petfalski et al. 1998; Sedger 2013), or viral RNA lariats (Galvis et al. 2017; Perng and Jones 2010; Plotch and Krug 1986; Ulfendahl et al. 1989), thereby controlling cell-intrinsic defense against intracellular virus replication. Inherited DBR1 deficiency probably underlies viral infection of the brainstem through the disruption of brainstem-specific and cell-intrinsic immunity to viruses, including HSV-1. However, the cellular basis of brainstem infection in patients with DBR1 mutations remains a matter of speculation, as iPSC-derived brainstem cells have not yet been generated and tested.
Concluding remarks
Human genetic studies of HSE have provided proof-of-principle that TLR3 and snoRNA31 are essential for cell-intrinsic immunity to HSV-1 in the forebrain, whereas DBR1 is essential for cell-intrinsic immunity to various viruses, including HSV-1, in the brainstem (Fig. 1). These observations pave the way for further studies of CNS tissue-specific, cell-intrinsic, as opposed to leukocyte-mediated, innate or adaptive immunity to HSV-1 in humans (Zhang et al. 2019). They also open up new perspectives for studies of inborn errors of immunity conferring predisposition to other types of isolated viral encephalitis, such as BVE due to IBV or norovirus (Zhang et al. 2018), or CNS infection due to varicella zoster virus (Ogunjimi et al. 2017). A number of unresolved questions remain: (1) is forebrain HSE physiologically homogeneous? The discovery of the role of snoRNA31 raises doubts about this, as no connection between this molecule and TLR3 or IFN has been established. It is possible that snoRNA31 is connected to TLR3-, IFN-inducible anti-HSV-1 immunity via hitherto unknown mechanisms. Alternatively, other TLR3- and IFN-independent molecular mechanisms may be involved in forebrain and/or brainstem HSE. (2) What is the basis of incomplete clinical penetrance for all the monogenic etiologies of forebrain HSE? This incomplete penetrance is consistent with the sporadic nature of HSE. Both host (age at infection, modifying genes) and viral factors (viral load, virus strain) may govern clinical penetrance. (3) Are there genetic etiologies, including monogenic lesions in particular, in the vast majority (~ 93%) of patients with forebrain or brainstem HSE for whom no genetic etiology has yet been identified? Genome-wide searches by both reverse and forward genetic approaches are underway. This work will build on the previous discoveries of HSE-causing genes, which will serve as anchor genes for the interpretation of genetic data. (4) Are there digenic or oligogenic inborn errors underlying HSE? Novel bioinformatic tools will be required to tackle this question. (5) Do CNS cells other than cortical neurons and oligodendrocytes contribute to host defense against HSV-1 in the brain? The development of novel hPSC-based protocols will be required to tackle this problem. (6) Is antiviral immunity in the CNS governed principally by anatomic region of the brain (e.g. BVE due to various viruses), causal virus (e.g. HSV-1 vs. other viruses), or both? Future studies will gradually define the human genetic and immunological architecture of childhood HSE, with both clinical and biological implications.

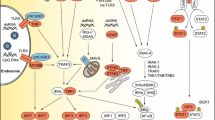
Inborn errors of immunity conferring predisposition to childhood HSE. Monogenic inborn errors of the TLR3-IFN circuit or snoRNA31 confer susceptibility to forebrain HSE, whereas inborn errors of DBR1 underlie brainstem HSE. TLR3 signaling is initiated by the recognition of dsRNA, inducing activation of the IRF3 and NF-κB pathways via TRIF, leading to the production of IFN-α/β and/or IFN-λ. Mutations of six TLR3 signaling pathway genes (TLR3, UNC93B1, TRIF, TRAF3, TBK1, and IRF3, highlighted in blue) have been found in patients with forebrain HSE. Mutations of other two genes of the TLR3-IFN circuit (NEMO, STAT1, in orange) have been found in patients suffering from HSE together with mycobacterial disease. Impaired TLR3-dependent, IFN-mediated cortical neuron- and oligodendrocyte-autonomous anti-HSV-1 immunity may underlie the pathogenesis of HSE in patients with TLR3 pathway gene defects. SnoRNA31 deficiency impairs cortical neuron-intrinsic immunity to HSV-1. DBR1 is a protein that shuttles between the cell nucleus and cytoplasm. DBR1 deficiency leads to defective RNA lariat metabolism and impaired cell-intrinsic immunity to HSV-1, despite normal cellular responses to stimulation with TLR3 or IFN-α/β. The fine molecular and cellular mechanisms by which snoRNA31 or DBR1 (in violet) deficiencies cause forebrain and brainstem HSE, respectively, remain to be dissected in detail
References
Abel L, Plancoulaine S, Jouanguy E, Zhang SY, Mahfoufi N, Nicolas N, Sancho-Shimizu V, Alcais A, Guo Y, Cardon A, Boucherit S, Obach D, Clozel T, Lorenzo L, Amsallem D, Berquin P, Blanc T, Bost-Bru C, Chabrier S, Chabrol B, Cheuret E, Dulac O, Evrard P, Heron B, Lazaro L, Mancini J, Pedespan JM, Rivier F, Vallee L, Lebon P, Rozenberg F, Casanova JL, Tardieu M (2010) Age-dependent mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J Pediatr 157:623–629. https://doi.org/10.1016/j.jpeds.2010.04.020
Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL (2010) Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity?. Ann N Y Acad Sci 1214:18–33. https://doi.org/10.1111/j.1749-6632.2010.05834.x
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L, Orntoft TF, Hornung V, Paludan SR, Mikkelsen JG, Fujita T, Christiansen M, Hartmann R, Mogensen TH (2015) Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 212:1371–1379. https://doi.org/10.1084/jem.20142274
Arenas J, Hurwitz J (1987) Purification of a RNA debranching activity from HeLa cells. J Biol Chem 262:4274–4279
Armangue T, Baucells BJ, Vlagea A, Petit-Pedrol M, Esteve-Sole A, Deya-Martinez A, Ruiz-Garcia R, Juan M, Perez de Diego R, Dalmau J, Alsina L (2019) Toll-like receptor 3 deficiency in autoimmune encephalitis post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm 6:e611. https://doi.org/10.1212/NXI.0000000000000611
Audry M, Ciancanelli M, Yang K, Cobat A, Chang HH, Sancho-Shimizu V, Lorenzo L, Niehues T, Reichenbach J, Li XX, Israel A, Abel L, Casanova JL, Zhang SY, Jouanguy E, Puel A (2011) NEMO is a key component of NF-kappaB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J Allergy Clin Immunol 128:610–617. https://doi.org/10.1016/j.jaci.2011.04.059
Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, Kidd JR, Kidd KK, Alcais A, Ragimbeau J, Pellegrini S, Abel L, Casanova JL, Quintana-Murci L (2009) Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet 5:e1000562. https://doi.org/10.1371/journal.pgen.1000562
Campbell M, Klapper PE, Longson M (1982) Acyclovir in herpes encephalitis. Lancet 1:38
Casanova JL (2015a) Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci USA 112:E7118–E7127. https://doi.org/10.1073/pnas.1521644112
Casanova JL (2015b) Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci USA 112:E7128–E7137. https://doi.org/10.1073/pnas.1521651112
Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL (2006) Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312
Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, Hawkins K, Casanova JL, Arkwright PD (2006) Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol 176:5078–5083
Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J, Zhang SY, Bustamante J, Vogt G, Lejeune J, Mayola E, de Beaucoudrey L, Abel L, Engelhard D, Casanova JL (2009) A partial form of recessive STAT1 deficiency in humans. J Clin Invest 119:1502–1514. https://doi.org/10.1172/JCI37083
Chapman KB, Boeke JD (1991) Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 65:483–492
Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL (2003) Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 33:388–391
Galvis AE, Fisher HE, Fan H, Camerini D (2017) Conformational changes in the 5′ end of the HIV-1 genome dependent on the debranching enzyme DBR1 during early stages of infection. J Virol. https://doi.org/10.1128/JVI.01377-17
Garcia-Morato B, Apalategi CA, Bravo-Galego L, Blaquez Moreno A, Soimon-Fuentes M, Garmendia J, Mendez Echevarria A, Del Rosal RT, Dominguez-Solo A, Lopez-Granados E, Reyburn H, Roriguez Pena R (2019) Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J Allergy Clin Immunol 144:309–312
Gazquez I, Jover A, Puig T, Vincente de Vera C, Rubio M (1996) Familial herpes encephalitis. Lancet 347:910
Gnann JW Jr, Whitley RJ (2017) Herpes simplex encephalitis: an update. Curr Infect Dis Rep 19:13. https://doi.org/10.1007/s11908-017-0568-7
Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, Philippe PB, Perez de Diego R, Cardon A, Vogt G, Picard C, Andrianirina ZZ, Rozenberg F, Lebon P, Plancoulaine S, Tardieu M, Valerie D, Jouanguy E, Chaussabel D, Geissmann F, Abel L, Casanova JL, Zhang SY (2011) Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med 208:2083–2098. https://doi.org/10.1084/jem.20101568
Han B, Park HK, Ching T, Panneerselvam J, Wang H, Shen Y, Zhang J, Li L, Che R, Garmire L, Fei P (2017) Human DBR1 modulates the recycling of snRNPs to affect alternative RNA splicing and contributes to the suppression of cancer development. Oncogene. https://doi.org/10.1038/onc.2017.150
Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY, Casanova JL (2012) Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. https://doi.org/10.1084/jem.20111316
Hjalmarsson A, Blomqvist P, Skoldenberg B (2007) Herpes simplex encephalitis in Sweden, 1990–2001: incidence, morbidity, and mortality. Clin Infect Dis 45:875–880
Jackson AC, Melanson M, Rossiter JP (2002) Familial herpes simplex encephalitis. Ann Neurol 51:406–407
Jacquier A, Rosbash M (1986) RNA splicing and intron turnover are greatly diminished by a mutant yeast branch point. Proc Natl Acad Sci USA 83:5835–5839
Jorjani H, Kehr S, Jedlinski DJ, Gumienny R, Hertel J, Stadler PF, Zavolan M, Gruber AR (2016) An updated human snoRNAome. Nucleic Acids Res 44:5068–5082. https://doi.org/10.1093/nar/gkw386
Jubelt B, Mihai C, Li TM, Veerapaneni P (2011) Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 11:543–552. https://doi.org/10.1007/s11910-011-0228-5
Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13:816–825. https://doi.org/10.1038/sj.cdd.4401850
Kelly D, Kroll JS (2004) Encephalitis–beyond aciclovir. Adv Exp Med Biol 549:177–183
Kiss AM (2004) Human box H/ACA pseudouridylation guide RNA machinery. Mol Cell Biol 24:11
Kohl S (1988) Herpes simplex virus encephalitis in children. Pediatr Clin North Am 35:465–483
Kohl S (1998) Herpes simplex virus. In: Feigin RD, Cherry JD (eds) Textbook of pediatric infectious diseases, vol 2, 4th edn. W.B. Saunders, Philadelphia, pp 1703–1732
Koskiniemi M, Saarinen A, Klapper PE, Sarna S, Cleator G, Vaheri A (1995) Familial herpes encephalitis. Lancet 346:1553
Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD (2012) Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773. https://doi.org/10.1038/nature11583
Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, Itan Y, Ewaleifoh O, Rapaport F, Carlile TM, Carter-Timofte ME, Paquet D, Dobbs K, Zimmer B, Gao D, Rojas-Duran MF, Kwart D, Rattina V, Ciancanelli MJ, McAlpine JL, Lorenzo L, Boucherit S, Rozenberg F, Halwani R, Henry B, Amenzoui N, Alsum Z, Marques L, Church JA, Al-Muhsen S, Tardieu M, Bousfiha AA, Paludan SR, Mogensen TH, Quintana-Murci L, Tessier-Lavigne M, Smith GA, Notarangelo LD, Studer L, Gilbert W, Abel L, Casanova JL, Zhang SY (2019) Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat Med 25:1873–1884. https://doi.org/10.1038/s41591-019-0672-3
Lerner AM, Levine DP, Reyes MP (1983) Two cases of herpes simplex virus encephalitis in the same family. N Engl J Med 308:1481
Liang F, Glans H, Enoksson SL, Kolios AGA, Lore K, Nilsson J (2019) Recurrent herpes zoster ophthalmicus in a patient with a novel toll-like receptor 3 variant linked to compromised activation capacity in fibroblasts. J Infect Dis. https://doi.org/10.1093/infdis/jiz229
Lim HK, Seppanen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, Dell W, Lorenzo L, Byun M, Pauwels E, Ronnelid Y, Cai X, Boucherit S, Jouanguy E, Paetau A, Lebon P, Rozenberg F, Tardieu M, Abel L, Yildiran A, Vergison A, Roivainen R, Etzioni A, Tienari PJ, Casanova JL, Zhang SY (2014) TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk. Neurology 83:1888–1897. https://doi.org/10.1212/WNL.0000000000000999
Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, Dobbs K, Hernandez N, Goudin N, Hasek ML, Garcia Reino EJ, Lafaille FG, Lorenzo L, Luthra P, Kochetkov T, Bigio B, Boucherit S, Rozenberg F, Vedrinne C, Keller MD, Itan Y, Garcia-Sastre A, Celard M, Orange JS, Ciancanelli MJ, Meyts I, Zhang Q, Abel L, Notarangelo LD, Snoeck HW, Casanova JL, Zhang SY (2019) Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med 216(9):2038–2056. https://doi.org/10.1084/jem.20181621
Mikami T, Miyashita H, Takatsuka S, Kuroki Y, Matsushima N (2012) Molecular evolution of vertebrate Toll-like receptors: evolutionary rate difference between their leucine-rich repeats and their TIR domains. Gene 503:235–243. https://doi.org/10.1016/j.gene.2012.04.007
Mork N, Kofod-Olsen E, Sorensen KB, Bach E, Orntoft TF, Ostergaard L, Paludan SR, Christiansen M, Mogensen TH (2015) Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun 16:552–566. https://doi.org/10.1038/gene.2015.46
Murray JL, Sheng J, Rubin DH (2014) A role for H/ACA and C/D small nucleolar RNAs in viral replication. Mol Biotechnol 56:429–437. https://doi.org/10.1007/s12033-013-9730-0
Nam K, Hudson RH, Chapman KB, Ganeshan K, Damha MJ, Boeke JD (1994) Yeast lariat debranching enzyme. Substrate and sequence specificity. J Biol Chem 269:20613–20621
Nam K, Lee G, Trambley J, Devine SE, Boeke JD (1997) Severe growth defect in a Schizosaccharomyces pombe mutant defective in intron lariat degradation. Mol Cell Biol 17:809–818
Niehues T, Reichenbach J, Neubert J, Gudowius S, Puel A, Horneff G, Lainka E, Dirksen U, Schroten H, Döffinger R, Casanova JL, Wahn V (2004) A NEMO-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immunol 114:1456–1462
Notarangelo L, Casanova JL, Conley ME, Chapel H, Fischer A, Puck J, Roifman C, Seger R, Geha RS (2006) Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005. J Allergy Clin Immunol 117:883–896
Ochs H, Smith CIE, Puck J (2007) Primary Immunodeficiencies: a molecular and genetic approach, 2nd edn. Oxford University Press, New York
Ogunjimi B, Zhang SY, Sorensen KB, Skipper KA, Carter-Timofte M, Kerner G, Luecke S, Prabakaran T, Cai Y, Meester J, Bartholomeus E, Bolar NA, Vandeweyer G, Claes C, Sillis Y, Lorenzo L, Fiorenza RA, Boucherit S, Dielman C, Heynderickx S, Elias G, Kurotova A, Auwera AV, Verstraete L, Lagae L, Verhelst H, Jansen A, Ramet J, Suls A, Smits E, Ceulemans B, Van Laer L, Plat Wilson G, Kreth J, Picard C, Von Bernuth H, Fluss J, Chabrier S, Abel L, Mortier G, Fribourg S, Mikkelsen JG, Casanova JL, Paludan SR, Mogensen TH (2017) Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest 127:3543–3556. https://doi.org/10.1172/JCI92280
Ooi SL, Samarsky DA, Fournier MJ, Boeke JD (1998) Intronic snoRNA biosynthesis in Saccharomyces cerevisiae depends on the lariat-debranching enzyme: intron length effects and activity of a precursor snoRNA. RNA 4:1096–1110
Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al-Muhsen S, Jouanguy E, Zhang SY, Abel L, Casanova JL (2010) Human TRAF3 adaptor molecule deficiency leads to impaired toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33:400–411. https://doi.org/10.1016/j.immuni.2010.08.014
Perng GC, Jones C (2010) Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415. https://doi.org/10.1155/2010/262415
Petfalski E, Dandekar T, Henry Y, Tollervey D (1998) Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol Cell Biol 18:1181–1189
Plotch SJ, Krug RM (1986) In vitro splicing of influenza viral NS1 mRNA and NS1-beta-globin chimeras: possible mechanisms for the control of viral mRNA splicing. Proc Natl Acad Sci USA 83:5444–5448
Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, Doffinger R, Bonnet M, Filipe-Santos O, Beaucoudrey L, Durandy A, Horneff G, Novelli F, Wahn V, Smahi A, Israel A, Niehues T, Casanova JL (2006) The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet 78:691–701
Raschilas F, Wolff M, Delatour F, Chaffaut C, De Broucker T, Chevret S, Lebon P, Canton P, Rozenberg F (2002) Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin Infect Dis 35:254–260
Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, Ulhoi BP, Holm TH, Mogensen TH, Owens T, Nyengaard JR, Thomsen AR, Paludan SR (2012) TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 122:1368–1376. https://doi.org/10.1172/JCI60893
Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vaegter C, Nyengaard JR, Fitzgerald KA, Paludan SR (2016) Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 7:13348. https://doi.org/10.1038/ncomms13348
Ruskin B, Green MR (1985) An RNA processing activity that debranches RNA lariats. Science 229:135–140
Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, Mahvelati F, Herman M, Ciancanelli M, Guo Y, Alsum Z, Alkhamis N, Al-Makadma AS, Ghadiri A, Boucherit S, Plancoulaine S, Picard C, Rozenberg F, Tardieu M, Lebon P, Jouanguy E, Rezaei N, Seya T, Matsumoto M, Chaussabel D, Puel A, Zhang SY, Abel L, Al-Muhsen S, Casanova JL (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121:4889–4902. https://doi.org/10.1172/JCI59259
Sato R, Kato A, Chimura T, Saitoh SI, Shibata T, Murakami Y, Fukui R, Liu K, Zhang Y, Arii J, Sun-Wada GH, Wada Y, Ikenoue T, Barber GN, Manabe T, Kawaguchi Y, Miyake K (2018) Combating herpesvirus encephalitis by potentiating a TLR3-mTORC2 axis. Nat Immunol 19:1071–1082. https://doi.org/10.1038/s41590-018-0203-2
Schlesinger Y, Buller RS, Brunstrom JE, Moran CJ, Storch GA (1995) Expanded spectrum of herpes simplex encephalitis in childhood. J Pediatr 126:234–241
Sedger LM (2013) microRNA control of interferons and interferon induced anti-viral activity. Mol Immunol 56:781–793. https://doi.org/10.1016/j.molimm.2013.07.009
Sironi M, Peri AM, Cagliani R, Forni D, Riva S, Biasin M, Clerici M, Gori A (2017) TLR3 mutations in adult patients with herpes simplex virus and varicella-zoster virus encephalitis. J Infect Dis 215:1430–1434. https://doi.org/10.1093/infdis/jix166
Smith G (2012) Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. https://doi.org/10.1146/annurev-micro-092611-150051
Smith MG, Lennette EH, Reames HR (1941) Isolation of the virus of herpes simplex and the demonstration of intranuclear inclusions in a case of acute encephalitis. Am J Pathol 17:55–68
Stahl JP, Mailles A (2019) Herpes simplex virus encephalitis update. Curr Opin Infect Dis 32:239–243. https://doi.org/10.1097/QCO.0000000000000554
Tangye SG, Al-Herz W, Bousfiha A, Hatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova JL, Sullivan KE (2020) Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. https://doi.org/10.1007/s10875-019-00737-x
Ulfendahl PJ, Kreivi JP, Akusjarvi G (1989) Role of the branch site/3′-splice site region in adenovirus-2 E1A pre-mRNA alternative splicing: evidence for 5′- and 3′-splice site co-operation. Nucleic Acids Res 17:925–938
Vitturi BK, Rosemberg S, Arita FN, da Rocha AJ, Forte WCN, Tilbery CP (2019) Multiphasic disseminated encephalomyelitis associated with herpes virus infection in a patient with TLR3 deficiency. Mult Scler Relat Disord 36:101379. https://doi.org/10.1016/j.msard.2019.101379
Whitley RJ (2002) Herpes simplex virus in children. Curr Treat Options Neurol 4:231–237
Whitley RJ (2006) Herpes simplex encephalitis: adolescents and adults. Antiviral Res 71:141–148
Whitley RJ (2015) Herpes simplex virus infections of the central nervous system. Continuum 21:1704–1713. https://doi.org/10.1212/CON.0000000000000243
Whitley RJ, Gnann JW (1993) The epidemiology and clinical manifestation of herpes simplex virus infections. In: Roizman RJW, Lopez C (eds) The humanherpesviruses. Raven Press, New York, pp 69–105
Zhang SY, Casanova JL (2015) Inborn errors underlying herpes simplex encephalitis: from TLR3 to IRF3. J Exp Med 212:1342–1343. https://doi.org/10.1084/jem.2129insight4
Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527
Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, Lafaille FG, Tsumura M, Cobat A, Luo J, Volpi S, Zimmer B, Sakata S, Dinis A, Ohara O, Garcia Reino EJ, Dobbs K, Hasek M, Holloway SP, McCammon K, Hussong SA, DeRosa N, Van Skike CE, Katolik A, Lorenzo L, Hyodo M, Faria E, Halwani R, Fukuhara R, Smith GA, Galvan V, Damha MJ, Al-Muhsen S, Itan Y, Boeke JD, Notarangelo LD, Studer L, Kobayashi M, Diogo L, Fairbrother WG, Abel L, Rosenberg BR, Hart PJ, Etzioni A, Casanova JL (2018) Inborn errors of RNA lariat metabolism in humans with brainstem viral infection. Cell 172:952–965. https://doi.org/10.1016/j.cell.2018.02.019
Zhang SY, Jouanguy E, Zhang Q, Abel L, Puel A, Casanova JL (2019) Human inborn errors of immunity to infection affecting cells other than leukocytes: from the immune system to the whole organism. Curr Opin Immunol 59:88–100. https://doi.org/10.1016/j.coi.2019.03.008
Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, Liu B, Tchieu J, Steinbeck JA, Lafaille F, Volpi S, Notarangelo LD, Casanova JL, Zhang SY, Smith GA, Studer L (2018) Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci USA 115:E8775–E8782. https://doi.org/10.1073/pnas.1809853115
Acknowledgements
I thank Jean-Laurent Casanova for critical reading of this manuscript. I thank the members of both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions, and technical and administrative assistance. I would also like to thank the patients and their families for participating in our studies. Our work on the human genetics of herpes simplex encephalitis was funded in part by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), Clinical and Translational Science Award (CTSA) program (UL1TR001866), NIH (R21NS084255, R01AI088364 and R01NS072381), the French National Research Agency (ANR) (ANR-14-CE14-0008-01, AAPG2018-SEAeHostFactors, AAPG2019-CNSVIRGEN and ANR-10-LABX-62-IBEID), the Thrasher Research Fund, the Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, and the St Giles Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, SY. Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Hum Genet 139, 911–918 (2020). https://doi.org/10.1007/s00439-020-02127-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02127-5