
Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is caused by inborn errors of IFN-γ immunity. Affected patients are highly and selectively susceptible to weakly virulent mycobacteria, such as environmental mycobacteria and Bacillus Calmette–Guérin vaccines. Since 1996, disease-causing mutations have been reported in 15 genes, with allelic heterogeneity leading to 30 genetic disorders. Here, we briefly review the progress made in molecular, cellular, immunological, and clinical studies of MSMD since the last review published in 2018. Highlights include the discoveries of new genetic etiologies of MSMD: autosomal recessive (AR) complete deficiencies of (1) SPPL2a, (2) IL-12Rβ2, and (3) IL-23R, and (4) homozygosity for TYK2 P1104A, resulting in selective impairment of responses to IL-23. The penetrance of SPPL2a deficiency for MSMD is high, probably complete, whereas that of IL-12Rβ2 and IL-23R deficiencies, and TYK2 P1104A homozygosity, is incomplete, and probably low. SPPL2a deficiency has added weight to the notion that human cDC2 and Th1* cells are important for antimycobacterial immunity. Studies of IL-12Rβ2 and IL-23R deficiencies, and of homozygosity for P1104A TYK2, have shown that both IL-12 and IL-23 are required for optimal levels of IFN-γ. These recent findings illustrate how forward genetic studies of MSMD are continuing to shed light on the mechanisms of protective immunity to mycobacteria in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD, Online Mendelian Inheritance in Man, OMIM 209950) is a rare group of inborn errors of immunity characterized by selective susceptibility to clinical disease caused by attenuated Mycobacterium bovis-Bacillus Calmette-Guérin (BCG) vaccines and environmental mycobacteria (EM), in otherwise healthy patients, with normal resistance to other microbes, in the absence of overt immunological abnormalities in routine evaluations (“idiopathic” infections) (Casanova et al. 1995; Casanova and Abel 2002; Poyhonen et al. 2019; Newport and Levin 1994). The first clinical report of idiopathic disseminated BCG infection (BCG-osis) following vaccination probably dates back to 1951 (Mimouni 1951). MSMD affects about 1/50,000 individuals worldwide (Rosain et al. 2019a). Mycobacterial illnesses may have a wide range of clinical manifestations, from localized to disseminated, and acute to chronic infections, and with immature or mature granulomas (Poyhonen et al. 2019; Rosain et al. 2019a; Bustamante et al. 2014). Macrophage activation syndrome may occur in rare cases, probably due to uncontrolled mycobacterial infection (Rosain et al. 2019a; Humblet-Baron et al. 2019). Mycobacterial diseases typically have a childhood onset, but may also begin in adulthood. Some patients display a spontaneous improvement with age, once the first mycobacterial episode has been controlled by antibiotics and/or IFN-γ, whereas others present recurrences or multiple mycobacterial infections. The more virulent M. tuberculosis has also caused disease in some MSMD patients (Boisson-Dupuis et al. 2015; Shabani et al. 2019). About half of MSMD patients are also particularly susceptible to non-typhoidal Salmonella, and these patients therefore have a broad spectrum of clinical diseases, ranging from gastroenteritis to septicemia and disseminated infection (Rosain et al.2018,2019a; Bustamante et al. 2014; Prando et al. 2013; Beaucoudrey et al. 2010; Jindal et al. 2019; MacLennan et al. 2004; Doffinger et al. 1999). A significant proportion of MSMD patients, mostly in the context of specific genetic etiologies, also suffer from mucocutaneous candidiasis (CMC). Viral infections, herpes virus in particular, have also been reported in some genetic etiologies. Other viral, bacterial, fungal, and parasitic infections have been reported in a few or even single patients (Rosain et al. 2019a; Bustamante et al. 2014). Standard hematological and immunological screening results for primary immunodeficiencies (PIDs) are generally normal in these patients with “idiopathic” infections (Casanova and Abel 2002).
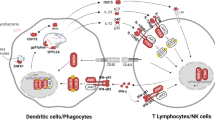
“Isolated” MSMD was the first group of PIDs characterized by a selective predisposition to one or a few infectious agents, the first “Mendelian infection”, to be deciphered at the molecular level, in 1996 (Table 1) (Casanova and Abel 2002; Newport et al. 1996; Jouanguy et al. 1996). This condition corresponds to the classic definition of MSMD. “Syndromic” MSMD was more recently defined as a combination of the mycobacterial infection phenotype with another, equally common phenotype, infectious or otherwise (e.g. type I interferonopathy) (Table 2). MSMD patients display considerable genetic heterogeneity. Mutations of 15 different genes (IFNGR1, IFNGR2, STAT1, JAK1, IRF8, SPPL2A, IL12B, IL12RB1, IL12RB2, IL23R, ISG15, TYK2, RORC, CYBB, and NEMO) cause isolated or syndromic MSMD (Tables 1, 2; Fig. 1) (Rosain et al. 2019a; Bustamante et al. 2014,2011; Boisson-Dupuis et al. 2018; Newport et al. 1996; Jouanguy et al. 1996,1999; Altare et al. 1998a,b; Fieschi et al. 2004; Vogt et al. 2005,2008; Chapgier et al. 2006,2009; Dupuis et al. 2001,2003; Filipe-Santos et al. 2006a; Kong et al. 2010a,b,2013,2018; Hambleton et al. 2011; Oleaga-Quintas et al. 2019; Kreins et al. 2015; Bogunovic et al. 2012; Okada et al. 2015; Eletto et al. 2016; Martinez-Barricarte et al. 2018). The mutations define 30 disorders, with high degrees of locus and genetic allelic heterogeneity, but with striking physiological homogeneity. Indeed, there is a common pathogenic mechanism to all these disorders: impairment of the production of, or responses to IFN-γ. These findings confirmed that IFN-γ, first described in 1965 as a pH-sensitive leukocytic antiviral IFN, is actually the macrophage-activating factor (MAF), as shown in 1983 by Carl Nathan (Nathan et al. 1983). The different genetic disorders are defined by the nature of the causal alleles (null or hypomorphic), protein levels (normal, low, or absent), and the mode of inheritance (recessive or dominant, autosomal or X-linked) (Rosain et al. 2019a; Bustamante et al. 2014). Various mutations, including single-nucleotide variants (SNVs) and structural variants (NV, including copy number variants (CNV)), have been reported in MSMD patients (Rosain et al. 2018,2019a,b; Bustamante et al. 2014; Humblet-Baron et al. 2019; Shabani et al. 2019; Jindal et al. 2019; Oleaga-Quintas et al. 2019; Pourakbari et al. 2019; Sarrafzadeh et al. 2019; Zhou et al. 2019). The molecular and clinical features of MSMD have been reviewed elsewhere (Rosain et al. 2019a; Bustamante et al. 2014; Filipe-Santos et al. 2006b; Haverkamp et al. 2014; Al-Muhsen and Casanova, 2008; Ramirez-Alejo and Santos-Argumedo 2014). Strikingly, the severity and penetrance of MSMD are inversely correlated with the residual levels of IFN-γ. Indeed, the only four genetic etiologies shown to date to be fully penetrant (i.e. Mendelian) in childhood and always lethal before the third decade of life in the absence of hematopoietic stem cell transplantation (HSCT) are complete IFN-γR1 and IFN-γR2 deficiencies (Bustamante et al. 2014). The other genetic etiologies of MSMD do not underlie bona fide MSMD, as mycobacterial disease does not segregate as a Mendelian trait in affected families. These monogenic disorders have incomplete penetrance due to residual IFN-γ activity. Overall, studies of MSMD have shown that human IFN-γ level is a quantitative trait that defines the outcome of mycobacterial infection. This review aims to provide a brief genetic, cellular, and clinical review of the four new etiologies of MSMD reported in 2018: complete deficiencies of AR SPPL2A, IL-12Rβ2, and IL-23, and homozygosity for TYK2 P1104A (Boisson-Dupuis et al. 2018; Martinez-Barricarte et al. 2018; Kong et al. 2018).

Number of patients with identified mutation in the genes involved in Mendelian susceptibility to mycobacterial disease (MSMD). Patients with isolated or syndromic MSMD have been included in this figure, indicating in the X axis the total number of patients according to the different deficiency idenfied in MSMD (Y axis). The figure included the total number of patients with genetic etiologies of MSMD published between 1996 and 2019
AR complete SPPL2a deficiency
SPPL2A encodes signal peptide peptidase-like 2 A (SPPL2a), an intramembrane protease of the GxGD protease family, the substrates of which include the N-terminal fragment (NTF) of the HLA invariant chain (CD74) expressed by HLA-class II+ antigen-presenting cells (Voss et al. 2013). Whole-exome sequencing (WES) and genome-wide linkage (GWL) analysis identified three patients with AR complete SPPL2a deficiency (Kong et al. 2018). These patients belong to two unrelated consanguineous kindreds from Morocco and Turkey. Clinical infectious disease caused by BCG has been reported in all three patients, in the absence of any other infections. Two different homozygous mutations affecting an essential splicing site, c.733+1G>A and c.1328-1G>A, lead to abnormal mRNA splicing, without detectable leakiness, in these patients (Kong et al. 2018). Overexpression studies revealed that the transcripts do not produce a protein (c.733+1G>A) or that they produced a truncated protein (c.1328-1G>A) (Kong et al. 2018). SPPL2a-deficient mice have impaired CD74 degradation in B lymphocytes and dendritic cells, and the development and function of both these cell subsets are affected (Schneppenheim et al. 2013; Beisner et al. 2013; Bergmann et al. 2013). By contrast, B-cell immunity seems to be intact in patients with SPPL2a deficiency. However, these patients have low frequencies of hematopoietic CD34+HLA-DR+ cells, like the corresponding mice, due to CD74 NTF toxicity (Bergmann et al. 2013). The accumulation of CD74 NTF impairs the development of dendritic cells type 2 (CD2), the major subset of circulating myeloid dendritic cells, which are also the IL-12/IL-23-producing CD1c+ CD11c+ cells. The counts and frequencies of peripheral T cells are normal, as is the distribution of the various subsets of memory CD4+ T cells, γδ T cells, and MAIT cells. However, this defect is associated with impaired IFN-γ production by memory T cells and CCR6+CXCR3+ memory Th cells, a Mycobacterium-specific memory subset referred to as Th1* cells (Kong et al. 2018). Patients with AR SPPL2a deficiency and autosomal dominant (AD) IRF8 deficiency display both a selective lack of cDC2 cells and a severe defect of IFN-γ production by CD4+ memory T cells and Mycobacterium-specific Th1* cells (Hambleton et al. 2011; Kong et al. 2018). These findings suggest that cDC2 cells are essential for the presentation of mycobacterial peptide antigens to CD4+ T cells, and the generation of Th1* cells, and that human cDC2 and/or Th1*cells are important for protective immunity to mycobacteria.
AR complete IL-12R2 deficiency
The receptor of IL-12 is a heterodimer of IL-12Rβ1 and IL-12Rβ2, whereas that of IL-23 is composed of IL-12Rβ1 and IL-23R (Presky et al. 1996; Parham et al. 2002). Biallelic mutations of IL12RB1 are the most frequent genetic cause of MSMD, and are found in about 60% of diagnosed patients (Rosain et al. 2018, 2019a; Beaucoudrey et al. 2010; Jindal et al. 2019; Pourakbari et al. 2019; Sarrafzadeh et al. 2019; Zhou et al. 2019; Vosse et al. 2013). Patients with IL-12Rβ1 deficiency suffer from mycobacterial diseases and salmonellosis, and from CMC due to impaired IL-23-dependent IL-17 production (Beaucoudrey et al. 2008). Asymptomatic individuals have been reported early on, attesting to the incomplete penetrance for MSMD, with only 50–70% of adults being symptomatic by the age of 40 years (Bustamante et al. 2014; Beaucoudrey et al. 2010; Esteve-Sole et al. 2018). Both IL-12 and IL-23 immunity are abolished in IL-12Rβ1 deficiency. A homozygous p.Q138* mutation was recently identified in the gene encoding the β2 subunit of the IL-12 receptor in a consanguineous Turkish family (Martinez-Barricarte et al. 2018). WES and GWL identified this genotype in a proband with disseminated BCG disease, and segregation analysis identified two other homozygous carriers: an uncle who suffered from disseminated tuberculosis (TB) at the age of five years and his younger asymptomatic brother. This pedigree is consistent with a low clinical penetrance for poorly virulent mycobacterial infections and predisposition to TB (Martinez-Barricarte et al. 2018). No CMC was reported in the three patients and IL-17 immunity was maintained. T-Saimiri T-cell lines had no detectable extracellular IL-12Rβ2 expression. In addition, no STAT4 phosphorylation was detected after stimulation of the patient’s T-saimiri cells with IL-12, whereas the response to IFN-α was normal. The cellular phenotype was complemented by retrotransduction with a wild-type copy of the IL12RB2 gene. The patients with IL-12Rβ2 deficiency have low frequencies of memory Th1 and Th1* cells, whereas their Th17 and Th2 cell levels are slightly low or normal. In the course of in vitro differentiation, IFN-γ production was abolished in Th1 conditions, as reported for IL-12Rβ1 deficiency but contrasting with IL-23R deficiency (discussed below). The identification and study of a kindred with AR complete IL-12Rβ2 deficiency showed that IL-12 is essential for immunity against mycobacteria, but only in a minority of patients. Indeed, penetrance for MSMD is incomplete, probably as low as 0.5%, because IL-23 can largely compensate for the loss of IL-12. Thus, both IL-12 and IL-23 play an important role in the control of MSMD.
AR complete IL-23R deficiency
A combination of WES and GWL analysis identified a homozygous mutation (p.C115Y) of IL23R, the gene encoding IL-23R (Martinez-Barricarte et al. 2018). The two siblings with this mutation had MSMD and were born to parents from a consanguineous Iranian kindred. Both had BCG disease, but neither had suffered from CMC. This mutant allele abolishes cellular responses to IL-23. In retrotransduction experiments on B-lymphocyte cell lines (B-LCLs, stably expressing STAT4), the mutant allele generates abundant mRNA, but only very small amounts of protein, and the proteins produced displayed abnormal N-glycosylation. In the patients’ EBV-B cell lines, STAT3 phosphorylation in response to IL-23 was abolished, and this defect was rescued by transduction with IL23R WT cDNA (Martinez-Barricarte et al. 2018). The IL-23R-deficient patients have normal frequencies of various leukocyte subsets, but low levels of MAIT cells. They also have abnormally low frequencies of memory Th1 and Th1* cells, and slightly low frequencies of Th17 and Th2 cells. The induction of Mycobacterium-specific CD4+ T (CCR6+ memory Th1*) cells led to lower levels of IFN-γ production in response to BCG and Mycobacterium tuberculosis in IL-12Rβ1-, IL-12Rβ2-, and IL-23R-deficient patients, a phenotype previously observed in ROR-γ/ROR-γT- and SPPL2A-deficient patients (Okada et al. 2015; Martinez-Barricarte et al. 2018; Kong et al. 2018). A computational genetic analysis showed that IL12RB1, IL12RB2, and IL23R, had evolved under similar levels of purifying selection (Martinez-Barricarte et al. 2018). The paucity of MSMD patients with IL-12Rβ2 or IL-23R deficiency is not therefore due to a greater rarity of loss-of-function (LOF) variants at these loci than at the IL12RB1 locus. Instead, it results from the lower clinical penetrance for MSMD, estimated at about 0.5%, consistent with the redundancy of lymphoid cell subsets producing IFN-γ and the partly overlapping roles of IL-12 and IL-23 in this process. Some lymphocytes respond better to IL-12 (ILC1, ILC2), some to IL-23 (ILC3), and still others respond equally well to both cytokines (MAIT, NKT), in terms of IFN-γ induction (Martinez-Barricarte et al. 2018; Lim et al. 2016). In the absence of one of these two cytokines (either IL-12 or IL-23), the other cytokine can partly compensate to ensure the production of IFN-γ. The characterization of the first human deficiency of IL-23R revealed the essential role of IL-23 in antimycobacterial immunity, at least in some individuals, most individuals being able to control mycobacteria in the absence of IL-23 (or IL-12). By contrast, IL-23 seems to be dispensable for Candida immunity in most patients, as in patients with IL-12Rβ1 deficiency (Beaucoudrey et al. 2010; Martinez-Barricarte et al. 2018).
Homozygosity for TYK2 P1104A
Non-receptor tyrosine-protein kinase (TYK2) is one of the four Janus kinases (JAKs). It is involved in signal transduction by four cytokine receptors, stimulated by IL-12, IL-23, IFN-α/β, and IL-10. Biallelic LOF mutations have been reported in patients with syndromic MSMD presenting both mycobacterial and viral infections (Kreins et al. 2015; Minegishi et al. 2006; Kilic et al. 2012). The peripheral leukocytes of the patients have impaired responses to IL-12 and IL-23, accounting for mycobacterial infections, as in patients with IL-12Rβ1 deficiency (Boisson-Dupuis et al. 2018). They also display impaired responses to IFN-α/β, accounting for the vulnerability of these patients to viral infections, albeit less severe than in patients with IFN-αR1 or IFN-αR2 deficiency (Hernandez et al. 2019; Duncan et al. 2015; Hoyos-Bachiloglu et al. 2017). Finally, the responses to IL-10 of these patients are poor, but stronger than those of patients with IL-10Rα or IL-10Rβ deficiencies, and, therefore, not associated with early-onset colitis (Kreins et al. 2015; Shouval et al. 2014). In this context, ten other patients with mycobacterial disease (three with MSMD and seven with TB) have been reported to be homozygous for a common missense variant (or polymorphism) of TYK2, P1104A (Boisson-Dupuis et al. 2018). The MSMD patients are from Iran, Sweden, and the USA, whereas the TB patients are from Algeria, Brazil, Chile, Morocco, and Turkey (Boisson-Dupuis et al. 2018). Penetrance was found to be much lower for MSMD (below 0.5%, everyone being exposed to weakly virulent mycobacteria) than for TB (greater than 50% in in areas of endemic TB, i.e. upon infection with M. tuberculosis, as discussed by Boisson-Dupuis S. in an accompanying review) (Kerner et al. 2019). The patients’ leukocytes respond poorly to IL-23 in terms of the induction of IFN-γ production. Homozygosity for P1104A affects IL-23 signaling in a selective manner, as cellular responses to IL-12, IFN-α/β, and IL-10 are intact in these patients. Homozygosity for P1104A TYK2 is a clinical phenocopy of IL-23R deficiency. Its cellular phenotype is very similar, but somewhat milder, as there are TYK2-independent, residual responses to each of the four cytokines, including IL-23. The patients displayed normal IL-17+ CD4+ T cell development ex vivo, consistent with the absence of CMC in these individuals. These patients are also normally resistant to other infectious diseases. This discovery demonstrates that common variants can underlie rare infectious diseases with low penetrance. Homozygosity for P1104A is present in as many as 1/600 humans of European descent, but the penetrance of this variant for MSMD is very low (no higher than 0.5%), and it is found in less than 0.5% of MSMD patients.
Conclusion
The identification and characterization of IL-12Rβ1 deficiency led to the discovery and characterization of IL-12Rβ2 and IL-23R deficiencies, and then homozygosity for P1104A TYK2. These findings collectively demonstrate the importance of IL-12- and IL-23-dependent IFN-γ immunity in humans, for optimal protection against mycobacteria. The penetrance for MSMD is about 50% when the immunity mediated by both cytokines is defective, whereas it is probably no higher than 0.5% when the immunity mediated by only one of these cytokines is defective. In most humans, IL-12 can compensate for the loss of IL-23, and vice versa. The cellular basis of the redundancy and synergy of these cytokines is that they stimulate only partly overlapping lymphocyte sets, with IL-12 stimulating preferentially ILC-1 and ILC-2, IL-23 preferentially stimulating MAIT and NKT cells, and both cytokines operating equally in CD4+ and CD8+ αβ T cells, γδ T cells and NK cells. Another original feature of P1104A TYK2 is its frequency in populations of European ancestry. Studies of SPPL2a deficiency have also helped to identify the cells involved in antimycobacterial immunity. Indeed, CD1c+ CD11c+ cells form the major subset of circulating myeloid dendritic cells contributing to the production of IL-12 and IL-23. They also seem to be important for the generation of Th1* cells. The discovery of these four new genetic etiologies of MSMD has important diagnostic and therapeutic implications. The molecular diagnosis of MSMD makes it possible to offer genetic counseling to affected families. Patients with these four disorders would also be predicted to benefit from IFN-γ therapy, in addition to antibiotics. Despite these recent discoveries, the genetic puzzle of mycobacterial infections remains far from completed, as no genetic etiology has yet been identified for almost half of all MSMD patients.
References
Al-Muhsen S, Casanova JL (2008) The genetic heterogeneity of Mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol 122(6):1043–1051
Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F et al (1998a) Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280(5368):1432–1435
Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S et al (1998b) Inherited interleukin 12 deficiency in a child with bacille Calmette–Guerin and Salmonella enteritidis disseminated infection. J Clin Invest. 102(12):2035–2040
Beisner DR, Langerak P, Parker AE, Dahlberg C, Otero FJ, Sutton SE et al (2013) The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J Exp Med 210(1):23–30
Bergmann H, Yabas M, Short A, Miosge L, Barthel N, Teh CE et al (2013) B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J Exp Med 210(1):31–40
Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D et al (2012) Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 337(6102):1684–1688
Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S et al (2015) Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev 264(1):103–120
Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G et al (2018) Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol 3:30
Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C et al (2011) Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 12(3):213–221
Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL (2014) Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol 26(6):454–470
Casanova JL, Abel L (2002) Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol 20:581–620
Casanova JL, Jouanguy E, Lamhamedi S, Blanche S, Fischer A (1995) Immunological conditions of children with BCG disseminated infection. Lancet 346(8974):581
Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A et al (2006) Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet 2(8):e131
Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J et al (2009) A partial form of recessive STAT1 deficiency in humans. J Clin Invest 119(6):1502–1514
de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M et al (2008) Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 205(7):1543–1550
de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J et al (2010) Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Med (Baltim) 89(6):381–402
Doffinger R, Jouanguy E, Altare F, Wood P, Shirakawa T, Novelli F et al (1999) Inheritable defects in interleukin-12- and interferon-gamma-mediated immunity and the TH1/TH2 paradigm in man. Allergy 54(5):409–412
Duncan CJ, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC et al (2015) Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med 7(307):154
Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J et al (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293(5528):300–303
Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S et al (2003) Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 33(3):388–391
Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F et al (2016) Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun 7:13992
Esteve-Sole A, Sanchez-Davila SP, Deya-Martinez A, Freeman AF, Zelazny AM, Dekker JP et al (2018) Severe BCG-osis misdiagnosed as multidrug-resistant tuberculosis in an IL-12Rbeta1-deficient Peruvian girl. J Clin Immunol 38(6):712–716
Fieschi C, Bosticardo M, de Beaucoudrey L, Boisson-Dupuis S, Feinberg J, Santos OF et al (2004) A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood 104(7):2095–2101
Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A et al (2006a) X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 203(7):1745–1759
Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J et al (2006b) Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 18(6):347–361
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J et al (2011) IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 365(2):127–138
Haverkamp MH, van de Vosse E, van Dissel JT (2014) Nontuberculous mycobacterial infections in children with inborn errors of the immune system. J Infect 68(Suppl 1):S134–S150
Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E et al (2019) Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med 216(9):2057–2070
Hoyos-Bachiloglu R, Chou J, Sodroski CN, Beano A, Bainter W, Angelova M et al (2017) A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J Clin Invest 127(12):4415–4420
Humblet-Baron S, Franckaert D, Dooley J, Ailal F, Bousfiha A, Deswarte C et al (2019) IFN-gamma and CD25 drive distinct pathologic features during hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol 143(6):2215–2226
Jindal AK, Suri D, Guleria S, Rawat A, Garg S, Bal A et al (2019) Recurrent Salmonella typhi infection and autoimmunity in a young boy with complete IL-12 receptor beta1 deficiency. J Clin Immunol 39(4):358–362
Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M et al (1996) Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med 335(26):1956–1961
Jouanguy E, Lamhamedi-Cherradi S, Lammas D, Dorman SE, Fondaneche MC, Dupuis S et al (1999) A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet 21(4):370–378
Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, Cobat A et al (2019) Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc Natl Acad Sci USA 116(21):10430–10434
Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L et al (2012) A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr 160(6):1055–1057
Kong XF, Ciancanelli M, Al-Hajjar S, Alsina L, Zumwalt T, Bustamante J et al (2010a) A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood 116(26):5895–5906
Kong XF, Vogt G, Chapgier A, Lamaze C, Bustamante J, Prando C et al (2010b) A novel form of cell type-specific partial IFN-gammaR1 deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Hum Mol Genet 19(3):434–444
Kong XF, Vogt G, Itan Y, Macura-Biegun A, Szaflarska A, Kowalczyk D et al (2013) Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet 22(4):769–781
Kong XF, Martinez-Barricarte R, Kennedy J, Mele F, Lazarov T, Deenick EK et al (2018) Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol 19(9):973–985
Kreins AY, Ciancanelli MJ, Okada S, Kong XF, Ramirez-Alejo N, Kilic SS et al (2015) Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med 212(10):1641–1662
Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L et al (2016) IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med 213(4):569–583
MacLennan C, Fieschi C, Lammas DA, Picard C, Dorman SE, Sanal O et al (2004) Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans. J Infect Dis 190(10):1755–1757
Martinez-Barricarte R, Markle JG, Ma CS, Deenick EK, Ramirez-Alejo N, Mele F et al (2018) Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol 3:30
Mimouni J (1951) Our experiences in three years of BCG vaccination at the center of the OPHS at Constantine; study of observed cases (25 cases of complications from BCG vaccination). Alger Med 55(8):1138–1147
Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S et al (2006) Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25(5):745–755
Nathan CF, Murray HW, Wiebe ME, Rubin BY (1983) Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 158(3):670–689
Newport M, Levin M (1994) Familial disseminated atypical mycobacterial disease. Immunol Lett. 43(1–2):133–138
Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R et al (1996) A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 335(26):1941–1949
Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M et al (2015) Immunodeficiencies: impairment of immunity to Candida and mycobacterium in humans with bi-allelic RORC mutations. Science 349(6248):606–613
Oleaga-Quintas C, Deswarte C, Moncada-Velez M, Metin A, Krishna Rao I, Kanik-Yuksek S et al (2019) A purely quantitative form of partial recessive IFN-gammaR2 deficiency caused by mutations of the initiation or second codon. Hum Mol Genet 28(3):524
Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J et al (2002) A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol 168(11):5699–5708
Pourakbari B, Hosseinpour Sadeghi R, Mahmoudi S, Parvaneh N, Keshavarz Valian S, Mamishi S (2019) Evaluation of interleukin-12 receptor beta1 and interferon gamma receptor 1 deficiency in patients with disseminated BCG infection. Allergol Immunopathol (Madr) 47(1):38–42
Poyhonen L, Bustamante J, Casanova JL, Jouanguy E, Zhang Q (2019) Life-threatening infections due to live-attenuated vaccines: early manifestations of inborn errors of immunity. J Clin Immunol 39(4):376–390
Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C et al (2013) Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Med (Baltim) 92(2):109–122
Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY et al (1996) A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci USA 93(24):14002–14007
Ramirez-Alejo N, Santos-Argumedo L (2014) Innate defects of the IL-12/IFN-gamma axis in susceptibility to infections by mycobacteria and salmonella. J Interferon Cytokine Res 34(5):307–317
Rosain J, Oleaga-Quintas C, Deswarte C, Verdin H, Marot S, Syridou G et al (2018) A variety of Alu-mediated copy number variations can underlie IL-12Rbeta1 deficiency. J Clin Immunol 38(5):617–627
Rosain J, Kong XF, Martinez-Barricarte R, Oleaga-Quintas C, Ramirez-Alejo N, Markle J et al (2019a) Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol Cell Biol 97(4):360–367
Rosain J, Deswarte C, Hancioglu G, Oleaga-Quintas C, Kutlug S, Kartal I et al (2019b) LINE-1-mediated AluYa5 insertion underlying complete autosomal recessive IFN-gammaR1 deficiency. J Clin Immunol 39(7):739–742
Sarrafzadeh SA, Nourizadeh M, Mahloojirad M, Fazlollahi MR, Shokouhi Shoormasti R, Badalzadeh M et al (2019) Molecular, immunological, and clinical features of 16 Iranian patients with mendelian susceptibility to mycobacterial disease. J Clin Immunol 39(3):287–297
Schneppenheim J, Dressel R, Huttl S, Lullmann-Rauch R, Engelke M, Dittmann K et al (2013) The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med 210(1):41–58
Shabani M, Aleyasin S, Kashef S, Zoghi S, Deswarte C, Casanova JL et al (2019) A novel recessive mutation of interferon-gamma receptor 1 in a patient with mycobacterium tuberculosis in bone marrow aspirate. J Clin Immunol 39(2):127–130
Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C et al (2014) Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol 122:177–210
van de Vosse E, Haverkamp MH, Ramirez-Alejo N, Martinez-Gallo M, Blancas-Galicia L, Metin A et al (2013) IL-12Rbeta1 deficiency: mutation update and description of the IL12RB1 variation database. Hum Mutat 34(10):1329–1339
Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C et al (2005) Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet 37(7):692–700
Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C et al (2008) Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med 205(8):1729–1737
Voss M, Schroder B, Fluhrer R (2013) Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim Biophys Acta 1828(12):2828–2839
Zhou X, Jia W, Ni Z, Wang A, Liu Z, Hou M et al (2019) Three novel compound heterozygous IL12RB1 mutations in Chinese patients with Mendelian susceptibility to mycobacterial disease. PLoS ONE 14(4):e0215648
Acknowledgements
I thank Jean-Laurent Casanova, Stéphanie Boisson-Dupuis, Jérémie Rosain, Anna-Lena Neehus and Laurent Abel for helpful discussions and support. I thank both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. The Laboratory of Human Genetics of Infectious Diseases is supported by the National Institute of Allergy and Infectious Diseases Grant no. 5R37AI095983, the Rockefeller University, the St. Giles Foundation, National Institute of Health and Medical Research (INSERM), Paris University, the Foundation for Medical Research (FRM), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency under the “Investments for the future” program (Grant no. ANR-10-IAHU-01) ANR-GENMSMD (ANR-16-CE17-0005-01 for JB), SRC 2017 (for JB) and ECOS Nord (C14S011 and C19S01-63407 for JB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bustamante, J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet 139, 993–1000 (2020). https://doi.org/10.1007/s00439-020-02120-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02120-y