
Abstract
Two hundred and one patients have been diagnosed with primary immunodeficiency diseases (PIDs) in our center from January 2004 to December 2009. The male-to-female ratio was 5.29:1. Spectrums of PIDs were as follows: predominantly antibody deficiency disease was the most common category (94 patients, 48.2%), followed by other well-defined immunodeficiency syndromes (40 patients, 20.5%), combined T and B cell immunodeficiencies (33 patients, 16.9%), congenital defects of phagocyte number and/or function (21 patients, 10.8%), and diseases of immune dysregulation (six patients, 3.1%). Agammaglobulinemia was the most frequent disease type. The median of diagnosis lag was 18.0 months. Pneumonia was the most common manifestation of PID patients. Some manifestations were prone to concentrate in certain diseases. As for therapy, 99 patients (50.8%) received intravenous immunoglobulin replacement therapy; 13 patients received hematopoietic stem cell transplantation and nine of them were still alive. In this study, we sought to describe and analyze the distribution, clinical features, and therapy methods of PIDs among children diagnosed in our country and to compare with reports from other countries and regions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiency diseases (PIDs) are a heterogeneous group of rare inherited conditions, caused by defects in the immune system, and characterized by an unusual increased susceptibility to infections and a predisposition to allergy, autoimmunity, and malignancy [1]. Since the first discovery of congenital agammaglobulinemia by Ogdeon Bruton in 1952, more than 150 different PIDs have been identified [2].
In terms of immunology research, PIDs are often quoted as “experiments of nature” in humans and represent a chief resource for deciphering the underlying mechanisms that control the development and function of the human immune system. In clinical practice, pediatric physicians have little knowledge about these relatively rare and complex diseases, which makes the situation even worse. Patients may suffer from long-term morbidity without proper treatment due to wrong diagnosis, and some may die eventually. Efficient methods are expected to be developed for timely diagnosis and treatment.
Collections of clinical data may help people recognize the features of these diseases. Databases or national online registries for PID cases have been established in many countries and regions, which helped improve the diagnosis level in those areas [1, 3–10].
Currently, no national registry of PIDs has been established in China, and only several separated centers published their outcomes mainly in Chinese literature. Their researches were either performed with small sample size or focusing on genetic analysis for specific disease types [9, 11–13]. Thus, we try to describe and analyze the distribution and clinical features of various PID disorders among children diagnosed over the past 6 years at the Immunological Division, Department of Pediatrics, Xinhua Hospital. Then we tried to enhance the awareness and knowledge of pediatric physicians on PIDs, to provide opportunities for further research on these disorders and finally to improve the quality of lives of patients with PIDs.
Methods
Patient Enrollment
During the period from January 2004 to December 2009, 201 patients have been diagnosed with PIDs in our center. Two patients diagnosed with transient hypogammaglobulinemia were excluded (This disorder is temporary, and the prognosis is good, with most children reaching normal antibody levels by 2 to 4 years old). Four of the patients were lost in follow-ups. Thus the final analysis results of this study came from 195 patients.
Xinhua Hospital is a university teaching hospital and its pediatric immunology unit is one of the biggest tertiary referral centers for PIDs in China. All of the patients were first diagnosed and treated in our center or referred from other departments in Xinhua Hospital or other hospitals following the three criteria:
-
1.
Recurrent or severe infections (two or more pneumonias within 1 year, two or more other organs’ infections within 1 year, and two or more abscesses or deep infections including septicemia)
-
2.
Abnormal lab examinations (complete blood count, serum immunoglobulin level, and lymphocyte subsets analysis) suggesting impairment of immune system function
-
3.
Family history of PIDs
Then, further diagnosis was established according to the WHO criteria and Primary Immunodeficiency Classification Committee of International Union of Immunological Societies (IUIS) [14]. An informed consent was obtained from each patient’s parent or guardian before enrollment in the study.
Data Collection
For each diagnosed patients, a questionnaire was completed by doctors in our center and confirmed by a second doctor. The questionnaire was designed into six sections: basic sociodemographic data (including the patients’ name, gender, date and place of birth, age at onset of symptoms, age at diagnosis of PID, age and cause of death, parental consanguinity, early death in blood relatives, family history of PID, vaccination, and allergic history), diagnosis (according to the eight categories classified by IUIS), disease histories and clinical presentation, complication, laboratory tests, and treatments.
Laboratory analysis were performed using standard techniques, including complete blood count, peripheral blood smear, erythrocyte sedimentation rate, measurement of serum immunoglobulins (IgG, IgA, IgM, and IgE), complement hemolytic activity (CH50), peripheral blood lymphocyte subsets analysis (CD3, CD4, CD8, CD19, and CD56/16 subsets). Besides, antibodies for special pathogens (such as EB virus, tetanus, or diphtheria) were examined. If necessary, dihydrorhodamine 123 dye testing and assessment of the expression of CD18/CD15 on neutrophils by flow cytometry were performed. DNA sequencing and mutation analysis in candidate chromosome regions were performed in patients to confirm diagnosis. Secondary immunodeficiencies (such as related to HIV infection, drugs, or other conditions) were ruled out by doing relative tests.
Statistical Analysis
Data in database were analyzed with SAS 8.02. The data here were not in Gaussian distributions; hence, median and range were used to present the characters of focused variables. Consanguinity was defined as relatives having common ancestors within three generations.
Results
Spectrum of PID Cases
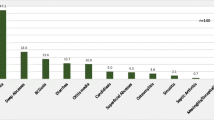
In this study, 195 patients were distributed in 23 diseases of five main categories of PIDs (Fig. 1). None of the patients was identified in any category of defects in the innate immunity, autoinflammatory disorders, or complement deficiencies.

Distribution of primary immunodeficiency disorders in Shanghai. N = 195, between 2004 and 2009, according to the IUIS classification
Predominantly antibody deficiency disease was the most common category (94 patients, 48.2%), followed by other well-defined immunodeficiency syndromes (40 patients, 20.5%), combined T and B cell immunodeficiencies (33 patients, 16.9%), congenital defects of phagocyte number and/or function (21 patients, 10.8%), and diseases of immune dysregulation (six patients, 3.1%). Nevertheless, one case cannot be classified into any kind of the eight categories (Table I).
Among the predominantly antibody deficiencies category, agammaglobulinemia (including Btk deficiency and non-Btk deficiency) was the most frequent phenotype. The most common phenotype manifested in other well-defined immunodeficiency syndromes category was Wiskott-Aldrich syndrome (WAS); T−B+ severe combined immunodeficiency (SCID) had the most patients in combined T and B cell immunodeficiency category; and chronic granulomatous disease (CGD) was more common than any other phenotype in congenital defects of phagocyte category (Table I).
Population Characteristics
The patient population comprised 164 boys and 31 girls, with a male-to-female ratio of 5.29:1. According to the data of the 5th National Census in 2000, general male-to-female ratios in Shanghai, Zhejiang, and Jiangsu Provinces, where most of the patients came from, are 1.06:1, 1.06:1, and 1.03:1, respectively [15]. Thus it implied that PIDs were generally prone to male patients. In the category of congenital defects of phagocyte, especial in the X-linked disorders such as CGD, the male-to-female ratio was much higher (9.5:1; Table I).
Age Distribution
In all patients, the median onset age of symptoms was 6 months (range, birth–180 months), the median age at diagnosis was 38 months (range, 13 days–246 months), and the median of the diagnosis lag, which represents the time elapsed between onset of symptoms and diagnosis, was 18 months (range, 2 days–225 months). Patients belonged to antibody deficiency category presented symptoms much later (median, 12.5 months). Especially, selective IgA deficiency and common variable immunodeficiency disorder (CVID) had the latest onset age of symptoms, 60 months and 36 months, respectively. And the diagnosis lag was also long in antibody deficiencies category, with average 66 months delay in hyper IgM syndrome (HIGM) and 60 months in CVID. At the time of diagnosis, 53 infants (27.2%) were below 1 year old, and 95% of the patients were under 15 years old (detailed in Table I). No antenatal diagnosis was made.
Geographic Distribution
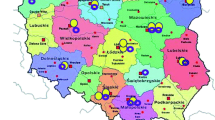
Figure 2 showed the geographic distribution of the patients diagnosed in our center. Among the 34 provinces in China, the origins of our patients covered 25 provinces, with only a few outermost regions being not included. Zhejiang, Jiangsu, Anhui Province, and Shanghai were representatives, which covered up to 59.0% (n = 115) of the total patients numbers. This distribution seemed to be consistent with the convenient degree to go to our hospital.

Geographic distribution of PID patients diagnosed in our center from 2004 to 2009. The map of China shows the whole 34 provinces. The labels and Arabic numerals represented the provinces where patients came from and the exact numbers of PID patients in that region classified into any kind of the eight categories
Consanguinity and Family History
The 195 patients came from 192 families. Of these patients, five were siblings from two families. Two sibs in the first family both belonged to antibody defect category, associated with decreased NK cells. The three patients in the second family were cousins, whose mothers were sisters. They were all diagnosed with X-linked agammaglobulinemia (XLAs). Neither of the two families was consanguinity family.
Four patients were from consanguinity families and other five patients’ parents had blood relationship within seven generations. Sixty-five patients (33.3%) had family histories of previous sib deaths at an early age. Among them, 33 died from severe infections and four of them were diagnosed with PIDs. Additionally, other four patients had family histories of PIDs.
Mortality
Twenty-four patients (12.3%) died during the study period (Table I). Eleven of them had family histories of previous sib death at an early age and one of the sibs died from PID. One patient was the descendant of consanguineous marriage and another patient’s parents had blood relationship within seven generations.
Among these 24 children, 13 were diagnosed with SCID, composing the largest part of death, which also suggesting SCID with the highest mortality rate. Severe infections (especially in lung and central nervous system), septicemia, and intracranial hemorrhage were the most common causes of death. Moreover, two patients developed malignancies. One patient with lymphoproliferative syndromes later suffered from non-Hodgkin lymphoma and died. The other patient diagnosed with CVID also suffered from non-Hodgkin lymphoma and died from severe infection during chemotherapy. Four patients diagnosed with SCID received hematopoietic stem cell transplantation (HSCT) but still died.
Clinical Manifestations of PIDs
Pneumonia topped the list of symptoms shown in PID patients, manifested by more than half of the patients (128 patients, 65.6%). Other commonly occurred symptoms were diarrhea, sinusitis, otitis media, lymphadenopathy, splenomegaly, eczema, bone and joint infections or arthritis, cutaneous infection, deep abscess, petechiae, hepatomegaly, and central nervous system infections (detailed in Table II). Almost all systems were involved. The respiratory system, gastrointestinal system, and immune organs were the three most affected systems or organs. One hundred and fifty patients (76.9%) had ear, nose, and throat (ENT) and airway infections. Seventy-seven patients (39.5%) had infections in more than one system or organ.
In different disease category, the clinical manifestations had their own characteristics. Pneumonia was most commonly presented in all categories. In addition to pneumonia, ENT infections, bone and joint infections, and diarrhea were most frequently seen in predominantly antibody deficiency diseases. As for other well-defined immunodeficiency syndromes (n = 40), petechiae (18 patients, 45.0%), diarrhea (15 patients, 37.5%), and eczema (15 patients, 37.5%) were more common. In patients with congenital defects of phagocyte number and/or function disease (n = 21), deep abscess (13 patients, 61.9%), lymphadenopathy (11 patients, 52.4%), and cutaneous infection (nine patients, 42.9%) were the most frequent symptoms. Some manifestations were prone to concentrate in certain diseases and the exact manifestation distributions were illustrated in Table II.
Associated Diseases
PID patients tended to have other immune system disorder complications. Among the 195 patients, we found 42 PID patients also exhibited allergic disease, autoimmune diseases, or malignancy.
Our cohort study showed that 35 patients (17.9%) in all had hypersensitivity diseases. Twenty-six of them had eczema or atopic dermatitis, nine patients had allergic history for specific food or drugs, four patients had asthma, and three patients had allergic rhinitis.
Nine patients caught complications as autoimmune diseases. One patient diagnosed with Omenn syndrome had systemic lupus erythematosus (SLE). One patient with XLA caught Guillain-Barre syndrome, and another one patient with CVID had autoimmune hepatitis. Six patients (one with XLA, two with HIGMs and three with CVIDs) had inflammatory bowel diseases (IBD). Besides, 25 patients had arthritis. Four of them were septic arthritis, and the other 21 patient’s arthritis seemed aseptic, whose symptoms resembled rheumatoid arthritis.
As to malignancies, seven patients in all gradually caught neoplasmas. Six patients (one with agammaglobulinemia, four with CVIDs, and one with lymphoproliferative syndromes) developed lymphoma and two of them died later. One patient diagnosed with non X-linked HIGM developed dysgerminoma.
About one quarter of the patients diagnosed with agammaglobulinemia, CVID and HIGM were accompanied with neutropenia (12/54, 6/27, and 3/11, respectively), most of which had compensatory elevated lymphocytes. Fifteen patients in antibody deficiency category presented anemia and ten with thrombocytopenia. Besides, 11 patients in all (diagnosed with DiGeorge syndrome, WAS, SCID, and XLA separately) had congenital heart disease histories. One patient with XLA simultaneously presented growth hormone deficiency. Another patient with XLA developed right leg paralysis after polio vaccine inoculation.
Therapy
Antibiotics were the most widely used treatments for PID patients. One hundred and fifty-eight patients (81.0%) used antibiotics. Ten patients received further anti-tuberculosis therapy.
Ninety-nine patients (50.8%) received intravenous immunoglobulin (IVIG) replacement therapy, no intramuscular or subcutaneous delivery. Of the patients, 60.6% (n = 57) were diagnosed with predominantly antibody deficiencies, composing the largest proportion treated with IVIG. IVIG were mainly used in the following five conditions: agammaglobulinemia (35 patients), SCID (16 patients), CVID (15 patients), WAS (15 patients), and HIGM (seven patients). No side effects of IVIG were recorded in these patients.
Thirteen patients received HSCT therapy, most of which belonged to SCID (five patients, 38.5%) and WAS (four patients, 30.8%). Other four patients were diagnosed with Chediak-Higashi syndrome (CHS; one patient), X-linked HIGM (two patients), and CGD (one patient). Four of the 13 patients died from sepsis and severe infections. Eight patients survived and did not require immunoglobulin supplementation. One WAS patient survived but still had sustained diarrhea and thrombocytopenia.
Other medications were used to alleviate symptoms. Thirteen out of 21 patients with defects of phagocyte had abscesses and needed surgeries for incision and drainage. Twenty-six patients were treated with immune regulators. For instance, interferon-α (100,000 U/kg, once a day, for 3 days, intramuscular injection) were used for antivirus therapy and interferon-γ-1b (50 μg/m2/day, if body surface area >0.5 m2, or 1.5 μg/m2/day, if body surface area ≤0.5 m2, three times a week, subcutaneous injection) for disorders of granulocyte respiratory burst activity, such as CGDs. Patients with persistent severe granulocytopenia, especially resulting from myelosuppression (WBC in peripheral blood <1,000/μl), were candidates for treatment with granulocyte-colony stimulating factor (G-CSF; 1–5 μg/kg, administrated subcutaneously each day, till WBC up to 10,000/μl). As for patients with cellular immunity dysfunction, thymosin alpha (1.6 mg, dissolved in 1.0 ml bi-distilled water for injection, twice a week, for 1 month, subcutaneous injection) was used to boost immune system (Table III).
Discussion
-
1.
Incidence
During 2004 to 2009, the annual average number of pediatric outpatients in Xinhua Hospital was 572,814 and one in every 2,850 is a PID patient. Our data represented a single-center study result and may underestimate the disease burden in our country, since it did not include patients admitted in other hospitals and those with milder forms of PID or quiet presentations. Patients with severe forms of PIDs that may die during infancy from severe infections before being identified as immunodeficiency patients were also excluded in this study.
Considering other national registries results, incidences of PIDs may present geographical or ethnic characters (Table IV). In Eastern Asia, European, Australia, and Latin America [3], incidences were lower, which were below five per 100,000 persons. While in Western Asia, the incidences were relatively high, over ten per 100,000 persons in Kuwait and six per 100,000 persons in Iran. This may be relevant to the high consanguineous marriage rate in these countries. The consanguinity rate in the general population was 22.6–42.1% in Kuwait [16] and 38.6% in Iran [17], while that in PID patients was 77% [5] and 68.5% [1] separately.
Table IV Comparisons with other PID registries In USA, no report of the total PIDs incidence was available. However, a national telephone survey of 10,000 households by the Immune Deficiency Foundation in 2005 yielded an estimated population prevalence of diagnosed PIDs at one in 1,200 persons, which was much higher than many “rare” diseases [18, 19]. No data for PIDs in Africa was reported.
-
2.
Disease constitution
As shown in Table I, the distribution of PIDs in our study had its unique characteristics. Similar to other national registries results, antibody deficiency was the predominant category of PIDs. However, agammaglobulinemia here was the most common form of PIDs, neither the CVID as reported in Iran [1], Australia [6], USA [18], and Europe [4], nor the selective IgA deficiency in Spain [20], Latin America [3], Eastern Saudi Arabia [10], and so on. As reported, the prevalence of CVID is from 1:30,000 to 1:50,000 [21]; and the prevalence of XLA is around 1:380,000 [22], much lower than that of CVID. In Erik Glocker’s review, symptoms in CVID occur with two major peaks of onset at 5–10 and 20–30 years [23]. For the second peak, patients were not defined as children and excluded from our study, thus it may decrease the percentage of CVID in our results.
As to selective IgA deficiency, the incidence was lower in Asian populations and most of IgA-deficient individuals (about 85%–90%) are asymptomatic [24]. Moreover, no routine screening program has been established for IgA deficiency. The frequency of this disease was low in our study, with only two patients being identified.
The frequencies of other PID categories in our results were similar to the data reported from European Society for Immunodeficiency (ESID) and other national registries, with minor differences showing geographical, ethnic, and genetic predisposition of certain PID diseases (Table IV).
-
3.
Age distribution
In our study, almost all of the patients were children. Only one patient came to our center at the age of 20.5 and diagnosed with CVID, however, symptoms appeared being revealed when he was 11 years old. Pediatric patients were the major concerns in many reports [9, 25]. Nevertheless, adult patients were also included in many other studies [1, 3, 26]. These results suggested that PIDs were no longer confined to a pediatric disease, which was further convinced by surveys conducted in Europe and USA. It was shown that only 67.71% of the patients were below 20 in Europe [27] and about a quarter (26%) was below 18 in the USA [18].
In different countries, average onset ages were mostly under 15 months (Table IV). Symptoms in patients with antibody deficiencies appeared much later (median, 12.5 months). One of the reasons may be that antibodies in infants, mainly passively acquired from the mothers, maintained above protective levels till 7 to 9 months after birth [28]. Combined immunodeficiencies and phagocyte defect diseases presented manifestations much earlier (median, 3 months and 1 month), which indicated more severe conditions and need to be excluded first when undergoing diagnosis.
There was a large gap between the onset of symptoms and the final diagnosis of the diseases. Compared with reports in Iran, Kuwait, and Egypt, the median diagnosis lag in our study (18 months) was shorter, which in fact was still long. For the most common diseases in our study (agammaglobulinemia, CVID, WAS, and SCID), the median time of delay were 43.5, 60, 4, and 2 months, respectively. These results compared well with previous studies on diagnostic delay. ESID data showed that the median diagnostic delay was 2 years (mean delay, 4.08 years) in Europe. As for the four diseases mentioned above, the mean diagnostic delay was 2.25, 5.23, 1.65, and 0.48 years [29]. The delay in diagnosis reflected the poor knowledge and low awareness about PIDs and led to significant morbidity which otherwise could been avoided if the diagnosis was established earlier. Recent progresses in recognition and diagnosis lead to a shorter tendency in the diagnosis lag.
The USA survey showed an unusual prolonged delay in diagnosis, which was even 12.4 years in 2007 [18]. This may partly associated with the large constitution of adult patients in America. Therefore, adult patients need to be taken into account in further research to gain an accurate description of the disease.
-
4.
Family history
Our result showed that 65 patients (33.3%) had family histories of previous sib deaths at an early age, and additional four patients had confirmed family history of PIDs. The proportion of patients with family histories was highest among those diagnosed with XLA (14 patients), SCID (ten patients), CGD (eight patients), and WAS (seven patients), which presented X-linked recessive heredity characters. And 83% of the families had records of early deaths in male family members. Thus, we inferred that most of the previous sib deaths may also attribute to immunodeficiency diseases. Inevitably, some early deaths irrelevant with PIDs could not be completely excluded in our statistics.
-
5.
Mortality
The mortality rate in our result (12.3%) is high, when compared with the ones reported in other countries, although we had a relatively shorter diagnosis lag. Some factors may be considered:
Generally speaking, shorter diagnosis lag means earlier diagnosis before severe symptoms appears and better prognosis. However, in our country, as doctors in primary hospitals were unfamiliar with these relatively rare and complex diseases, patients with mild symptoms would probably not be recognized and referred to specialist physicians. Considerable portion of patients diagnosed in our center always had severe infections or organ injuries and could not receive HSCT, which partly attribute to the high mortality. The median age of death in this study was 6 months. Symptoms appeared even earlier. The young onset and death age may indicate the severe conditions.
In addition, some of the diagnosed patients could not afford the expensive cost and gave up receiving treatments. Thus, the social–economical conditions can also affect the disease prognosis.
-
6.
Clinical presentations
Immunodeficiency is a failure to achieve immune function to provide efficient, self-limited host defense against the biotic and abiotic environment while preserving tolerance to self [30]. So it is always associated with a variety of clinical presentations. These presentations show distinct properties in different disease category and are the best reflections of the underlying physiologic effect of any deleterious genotype [30].
Our results were as follows:
-
Mucosa-related infections (such as recurrent sinusitis, otitis media, and diarrhea) were mainly found in antibody deficiency disease.
-
Arthritis commonly distributed in antibody deficiency diseases, especially in agammaglobulinemia (16 out of 54 patients, 29.6%). Monoarthritis and polyarthritis were nearly the same (12 vs. 13 patients).
The reported incidence of arthritis in XLA was 7–16.7% among Caucasians [22, 31] and a little higher in Asian population (29.0% in Hong Kong results [12] and 26.3% in Korean cohort [32]), which were consistent with our result.
-
Antibody deficiency patients (especially CVID patients) were at a higher risk of developing autoimmune diseases and malignancies.
-
Lymphoproliferation were frequently observed in antibody deficiency and phagocyte deficiency diseases.
-
Fungi infections were frequently distributed in patients with combined immune deficits and phagocytic disorders.
A clinical Working Party of ESID proposed a protocol for screening PID patients starting from clinical presentations. They classified the clinical presentations into eight patterns, matched each pattern to some special series of PID diseases, and initiated the diagnosis and therapy pathways [33]. Our results showed concentrate trends of manifestations in certain diseases and were consistent with theirs. Analyzing relationship between clinical presentations and disease types will facilitate early and efficient identification of PID within the large pool of candidate diseases.
-
-
7.
Complications
In our study, 26 PID patients had atopic dermatitis or eczema. Gu Heng et al. [34] reported that the standardized prevalence of atopic dermatitis in children in ten cities of China was proximately 3.07%, and the prevalence in Shanghai was 2.78%. In our study, the frequency of this complication was 13.3%, much higher than that in general pediatric population in Shanghai.
It was reported that the prevalence rates of SLE generally range from 20 to 70 per 100,000 person-years [35]. In Taiwan, the prevalence of childhood SLE was estimated as 6.3 per 100,000 [36]. In our study, we found one patient with SLE out of the total 195 patients (0.51%), the frequency of which was also much higher than that in Taiwan result.
IBD is also a type of autoimmune diseases [37, 38]. Six patients in our study had IBDs and all of them belonged to the predominantly antibody deficiency category. Available data suggested that the incidence of ulcerative colitis in Asian countries ranges from 0.4 to 2.1 per 100,000 population and the prevalence rates ranged from six to 30 per 100,000 population [39]. As for children, the annual incidence rates of IBD in UK and North America were estimated to be 5.2 to 7.05 per 100,000 pediatric populations in recent reports [40, 41]. Compared to these reports, the frequency of IBD (3.08%) in our study was higher than that in general population.
These data seemed to indicate increased prevalence rates of allergic and autoimmune disorders in PID patients in childhood. Moreover, among the nine patients who developed autoimmune diseases, four of them belonged to CVID (three with IBDs and one with autoimmune hepatitis). A large retrospective study concerning CVID patients showed that 52 out of 248 patients (21%) developed autoimmune diseases [42]. Other reports together indicated that approximately 20–40% of CVID patients developed manifest autoimmune diseases [43, 44]. Our result (four out of the 28 CVID patients, 14.3%) was close to the inferior limit of the reported range.
As for the seven patients accompanied with malignancies, six of them developed lymphoma, with four patients already diagnosed with CVID (2.05%). As reported, CVID patients were at a higher risk of developing malignancies. The most common malignancies were lymphoma and gastric cancer [42, 45]. Helen Chapel et al. [46] reported that about 2–8% of subjects with CVID were diagnosed with a non-Hodgkin lymphoma, which was consistent with our results. Gastric cancers were not found in our study yet.
-
8.
Therapy
Although improved supportive care and utilization of IVIG have extended the life span of PID patients, definitive cure is generally only achieved by HSCT currently. In our study, the 13 patients receiving HSCT belonged to SCID, WAS, CHS, X-linked HIGM, and CGD. This disease spectrum treated by transplantation was consistent with that reported in other countries [47, 48]. Currently, the stem cell donor can be an HLA-identical sibling (RID), a T-cell-depleted haplocompatible family member (MMRD), an HLA-matched unrelated donor (MUD), or cord blood unit and studies indicated that patients with RID and MUD had better survival and long-term reconstitution results [48, 49]. Early diagnosis, HSCT at early stage, careful supportive therapy, and monitoring for various pathogens were significant determinants for the successful transplantation therapy [47, 48] and hence, providing opportunities to improve the life qualities of PID patients.
In conclusion, our cohort represents a single-center sample of children from 25 provinces distributed in 23 diseases of five main categories of PIDs, with high frequency of antibody deficiency diseases. It may promote to establish a national registry of PID in China. Such registry will not only help to estimate the incidence and prevalence of PIDs but also to increase the awareness of physicians to PIDs, and to provide efficiency standardized diagnosis and therapy procedures for these disorders in the country.
Abbreviations
- CGD:
-
Chronic granulomatous disease
- CHS:
-
Chediak-Higashi syndrome
- CVID:
-
Common variable immunodeficiency disorder
- ESID:
-
European society for immunodeficiency
- HIGM:
-
Hyper-IgM syndrome
- HSCT:
-
Hematopoietic stem cell transplantation
- IBD:
-
Inflammatory bowel disease
- IUIS:
-
International Union of Immunological Societies
- IVIG:
-
Intravenous immunoglobulin
- PID:
-
Primary immunodeficiency disease
- SCID:
-
Severe combined immunodeficiency
- SLE:
-
Systermic lupus erythematosus
- WAS:
-
Wiskott-Aldrich syndrome
- XLA:
-
X-linked agammaglobulinemia
References
Rezaei N, Aghamohammadi A, Moin M, et al. Frequency and clinical manifestations of patients with primary immunodeficiency disorders in Iran: update from the Iranian Primary Immunodeficiency Registry. J Clin Immunol. 2006;26:519–32.
Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–94.
Leiva LE, Zelazco M, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol. 2007;27:101–8.
Gathmann B, Grimbacher B, Beaute J, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11.
Al-Herz W. Primary immunodeficiency disorders in Kuwait: first report from Kuwait National Primary Immunodeficiency Registry (2004–2006). J Clin Immunol. 2008;28:186–93.
Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–24.
Lim DL, Thong BY, Ho SY, et al. Primary immunodeficiency diseases in Singapore—the last 11 years. Singap Med J. 2003;44:579–86.
Hayakawa H, Iwata T, Yata J, Kobayashi N. Primary immunodeficiency syndrome in Japan. I. Overview of a nationwide survey on primary immunodeficiency syndrome. J Clin Immunol. 1981;1:31–9.
Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J Clin Immunol. 2005;25:162–73.
Al-Attas RA, Rahi AH. Primary antibody deficiency in Arabs: first report from eastern Saudi Arabia. J Clin Immunol. 1998;18:368–71.
Zhan YZ, Yang XQ. Clinical investigation of 135 primary immunodeficiency disorders in children. Chin J Pract Pediatr. 2009;24:132–4.
Lee PP, Chen TX, Jiang LP, et al. Clinical characteristics and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J Clin Immunol. 2010;30:121–31.
Lee PP, Lau YL. Primary immunodeficiencies: “new” disease in an old country. Cell Mol Immunol. 2009;6:397–406.
Notarangelo LD, Fischer A, Geha RS, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78.
National Bureau of Statistics of China. The 5th National Consus. http://www.stats.gov.cn/. 2001
Radovanovic Z, Shah N, Behbehani J. Prevalence and social correlates to consanguinity in Kuwait. Ann Saudi Med. 1999;19:206–10.
Saadat M, Ansari-Lari M, Farhud DD. Consanguineous marriage in Iran. Ann Hum Biol. 2004;31:263–9.
Primary immunodeficiency disease in America: 2007, the third national survey of patients.
Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497–502.
Matamoros FN, Mila LJ, Espanol BT, Raga BS, Fontan CG. Primary immunodeficiency syndrome in Spain: first report of the National Registry in Children and Adults. J Clin Immunol. 1997;17:333–9.
Herriot R, Sewell WA. Antibody deficiency. J Clin Pathol. 2008;61:994–1000.
Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85:193–202.
Glocker E, Ehl S, Grimbacher B. Common variable immunodeficiency in children. Curr Opin Pediatr. 2007;19:685–92.
Yel L. Selective IgA deficiency. J Clin Immunol. 2010;30:10–6.
Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol. 2009;29:343–51.
Primary Immunodeficiency Database in Japan. http://pidj.rcai.riken.jp/public_shoureisu.html.
Ballow M. Primary immunodeficiency disorders: antibody deficiency. J Allergy Clin Immunol. 2002;109:581–91.
Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004–06. Clin Exp Immunol. 2007;147:306–12.
Casanova JL, Fieschi C, Bustamante J, et al. From idiopathic infectious diseases to novel primary immunodeficiencies. J Allergy Clin Immunol. 2005;116:426–30.
Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol. 2002;104:221–30.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J. 2008;49:28–36.
de Vries E. Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. 2006;145:204–14.
Gu Heng YL, Yongshen L: Survey on the prevalence of childhood atopic dermatitis in ten cities of China. Chinese Journal of Dermatology 37:29–31, 2004
Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257–68.
Huang JL, Yao TC, See LC. Prevalence of pediatric systemic lupus erythematosus and juvenile chronic arthritis in a Chinese population: a nation-wide prospective population-based study in Taiwan. Clin Exp Rheumatol. 2004;22:776–80.
McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23.
Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–33.
Ooi CJ, Fock KM, Makharia GK, et al. The Asia-Pacific consensus on ulcerative colitis. J Gastroenterol Hepatol. 2010;25:453–68.
Kugathasan S, Judd RH, Hoffmann RG, et al. Epidemiologic and clinical characteristics of children with newly diagnosed inflammatory bowel disease in Wisconsin: a statewide population-based study. J Pediatr. 2003;143:525–31.
Sawczenko A, Sandhu BK, Logan RF, et al. Prospective survey of childhood inflammatory bowel disease in the British Isles. Lancet. 2001;357:1093–4.
Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48.
Salzer U, Maul-Pavicic A, Cunningham-Rundles C, et al. ICOS deficiency in patients with common variable immunodeficiency. Clin Immunol. 2004;113:234–40.
Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–16.
Gompels MM, Hodges E, Lock RJ, et al. Lymphoproliferative disease in antibody deficiency: a multi-centre study. Clin Exp Immunol. 2003;134:314–20.
Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–27.
Dvorak CC, Cowan MJ. Hematopoietic stem cell transplantation for primary immunodeficiency disease. Bone Marrow Transplant. 2008;41:119–26.
Tsuji Y, Imai K, Kajiwara M, et al. Hematopoietic stem cell transplantation for 30 patients with primary immunodeficiency diseases: 20 years experience of a single team. Bone Marrow Transplant. 2006;37:469–77.
Grunebaum E, Mazzolari E, Porta F, et al. Bone marrow transplantation for severe combined immune deficiency. JAMA. 2006;295:508–18.
Lee WI, Jaing TH, Huang JL. Distribution, infections, treatments and molecular analysis in a large cohort of patients with primary immunodeficiency diseases (PIDs) in Taiwan. J Clin Immunol. 2006;26:274–83.
The French PID study group. The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010;135:264–72.
Acknowledgments
We thank all the patients and their families for their kindness for permission, and we also thank their physicians who first treated and then referred them to our center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, LL., Jin, YY., Hao, YQ. et al. Distribution and Clinical Features of Primary Immunodeficiency Diseases in Chinese Children (2004–2009). J Clin Immunol 31, 297–308 (2011). https://doi.org/10.1007/s10875-010-9493-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-010-9493-3