
Abstract
Introduction
Despite rapid developments in the science of primary immunodeficiency diseases (PID), population characteristics and the burden of disease are poorly characterized. Aggregated data on PID via patient registries are a key component of the public health response. The web-enabled Australasian Society of Clinical Immunology and Allergy PID Register was designed and implemented to address gaps in knowledge of PID.
Methods
The register provided a cumulative, cross-sectional survey of PID patients in Australia and New Zealand via an online, single time point, center-based, voluntarily recalled, and patient-consented questionnaire.
Result
Eighty-eight centers reported 1,209 patients across 56 separate PID syndromes. The study prevalence (cases per 100,000 population) was 5.6 for Australia, 12.4 for the state of South Australia, and 4.9 for Australia and New Zealand combined. Predominately antibody deficiency syndromes accounted for 77% of patients. Common variable immunodeficiency was the most common diagnosis. Patients were geographically dispersed with 80% of centers reporting caseloads of less than 20 patients. Potentially preventable complications of disease were common. Immunoglobulin replacement therapy was used in 30 conditions with 26.5% of the total recipients having antibody deficiency disorders with normal serum IgG.
Conclusion
PID in Australia and New Zealand are prevalent, clinically diverse, geographically dispersed, and are characterized by high rates of potentially preventable morbidity and resource utilization. A public health focus on PID is required, including strategies to correct disparities in access to care, improve molecular diagnostics and reduce preventable complications of disease. Further studies in antibody deficiency syndromes with normal serum IgG are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Scientific discovery in the field of primary immunodeficiencies is progressing in advance of improvements in health service delivery to affected individuals [1]. Challenges include the broad range of diseases, clinical variations in presentation, complexity of laboratory and genetic assessment, and costs of therapy [1]. Gathering of information about primary immunodeficiency diseases (PID) through international PID registries is regarded as a fundamental component of recommendations to address the needs of patients and healthcare providers [1, 2]. Registry data provide detailed insight into the burden of disease and can be used to guide clinical trials, basic research, and service planning [2].
The spectrum of PID syndromes is now well defined. The International Union of Immunological Societies has classified approximately 120 separate PIDs with the molecular mechanism identified in more than 100 of these [3]. However, those disorders with unknown pathogenesis comprise the most prevalent conditions, including common variable immunodeficiency (CVID), IgG subclass deficiency, IgA deficiency, and selective antibody deficiency, and consensus case definitions for many of these syndromes are lacking. Furthermore, medical and community awareness of PID is unsatisfactory, with delayed diagnosis, preventable morbidity, and disparities of care commonplace [2].
To address these multifaceted needs in PID, a web-enabled patient register was designed and implemented under the auspices of the Australasian Society of Clinical Immunology and Allergy (ASCIA). The stated objective of the register is “To collect and analyze data on all patients with PID in Australasia to facilitate diagnosis, treatment, research, education, and quality assurance for patients with PID and their health care providers, and to guide the use of immunoglobulin replacement resources.” This report summarizes data collected and analyzed up until December 2006, and extends the single previous publication characterizing the Australian PID population [4].
Methods
The ASCIA PID register is an interactive, web-enabled Oracle database providing a cumulative cross-sectional survey of all PID patients in Australia and New Zealand (Fig. 1). The register was launched in October 2003 and New Zealand commenced registrations in 2004. The database was designed and administered by the authors (see Acknowledgements) and hosted by the George Institute for International Health. Access is provided to approved users via password control at http://www.immunodeficiency.org.au. Approved user status is available to healthcare professionals working in the field of PID subject to completion of a signed approved user agreement. The register holds single time point, center-based, voluntarily recalled, and patient-consented data. Nominal user incentive schemes and a network of center-based “PID officers” were established to mitigate the potential effects of recall and nonresponder biases.

Structure and design of ASCIA PID register. APSU = Australian Paediatric Surveillance Unit, ASCIA = Australasian Society of Clinical Immunology and Allergy.
Prevalence calculations were based on an Australian population of 20.6 million [5] and a New Zealand population of 4.1 million [6]. Aggregated data on intravenous immunoglobulin (IVIg) distribution for PID in Australia was obtained from the National Blood Authority, the agency responsible for national IVIg supply. Comparison between data on IVIg use in CVID and IgG subclass deficiency from both the register and from national supply data were used to approximate the target population (denominator) of IVIg recipients for these indications. These data were then extrapolated to the entire PID register population to derive measures of reporting and adjusted estimates of prevalence. Prevalence calculations apply to the study period only, and age and IVIg utilization data are dependent upon the assumption that patients survive, remain local, continue therapy, and do not recover from their PID.
The data acquisition process involved both Internet-based registrations into the PID register and the electronic merging of three previous databases (Australian Paediatric Surveillance Unit data, the original ASCIA PID survey [4] and an extension survey) (Fig. 1), with data collection beginning in 1990. In contrast to a number of other PID registries, data were not based on separate disease-specific surveys, enabling greater cross-sectional representation. The diagnosis of the patient was selected exclusively by the approved user from a drop-down list of PID diagnoses. Accuracy of diagnosis was the responsibility of the specialist in charge of the case. Details of the referring physician were collected. Data acquisition was a two-stage process. The first stage consisted of a standard questionnaire. The second stage questionnaire was tailored to the specific diagnostic group entered in stage one. Patient data was collected via code (name code, date of birth, residential postcode, and sex), enabling deletion of duplicate registrations. Data on ethnicity was not collected.
All aspects of the operation of the register are governed by comprehensive Human Research Ethics Committee approval (Sydney South West Area Health Service, Royal Prince Alfred Hospital zone, Australia, and Auckland Ethics Committee, New Zealand) and ethics consultation throughout Australia and New Zealand.
Results
Population Characteristics
The ASCIA PID register held data on 1,209 patients with PID from 88 participating centers across Australia and New Zealand on December 4, 2006. Fifty-six diagnoses were recorded. Of the 1,209 patients, 648 were male (53.6%) and 552 were female (45.7%) with the sex not recorded in 9 cases (Table I). The median age calculated from date of birth was 31 years (range: infancy to 96 years). A family history of the same or a similar disorder was positive in 211 of 677 patients where data was available (31.2%) and varied according to diagnosis as expected (Table I).
Diagnostic Groups
Single diagnoses were aggregated into six diagnostic groups with diagnoses of predominately antibody deficiency being the most common (77.0%). Patients were registered across all six groups. Common variable immunodeficiency was the single most common diagnosis with 464 patients (38.4% of total cases, Table I). Patients with CVID had a mean pretreatment IgG level of 4.1 g/l (SD 2.26) whereas patients with agammaglobulinemia reported a mean IgG of 1.1 g/l (SD 1.55).
Antibody deficiency syndromes characterized by normal serum total IgG (IgG subclass deficiency, specific antibody deficiency, or IgA deficiency) are among the least well characterized and most controversial of PID [7, 8]. A total of 364 patients (39.1% of antibody disorders and 30.1% of total PID registrations) with these disorders were registered, with IgG subclass deficiency the most common (n = 234, 25.1% of antibody deficiency patients, 19.4% of total patients, Table II).
Primary immunodeficiency diseases other than antibody deficiency syndromes comprised only 23% of total patients with hereditary angioedema (HAE, 55 patients, 4.5% of total), T-B− severe combined immunodeficiency (SCID) (41 patients, 3.4% of total), chronic granulomatous disease (33 patients, 2.7% of total), and hyper-IgM syndromes (25 patients, 2.1% of total) occurring most frequently (Table I).
The molecular basis of many PID have now been defined [3, 9]. Information about whether the diagnosis was established or confirmed by molecular genotyping was recorded in 583 PID patients (48.2% of total). However, genotypic diagnosis was established in only 32 of these 583 cases (5.5%).
Geographic Distribution and Prevalence Estimates
The geographic distribution of patients is a key limiting factor in provision of specialist services. Registrations were dispersed between 88 hospital or clinic centers across Australia and New Zealand. Whereas two major pediatric centers accounted for 20.1% of total cases, 70 centers (80% of total centers) had less than 20 patients registered, indicating limited caseloads at most centers.
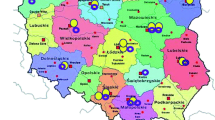
Although the majority of cases were registered in the most populous Australian states of New South Wales (39%) and Victoria (21%) (Fig. 2a), the population adjusted prevalence of PID varied widely between states and territories of Australia and New Zealand. Several states in Australian and New Zealand were greatly underrepresented (no registrations were recorded from the Northern Territory of Australia compared to 12.4 registrations per 100,000 in South Australia, Fig. 2b). Although there was regional variation in reporting, the profile of patients by diagnostic group was similar across all regions. In general, levels of underreporting correlated with population-adjusted clinical immunology workforce numbers in that area [10]. Overall, the observed Australian PID prevalence was 5.6 per 100,000 population, which is substantially greater than 2.1 per 100,000 population as reported in 1997 [4] (Table II). A prevalence of 4.9 per 100,000 population is observed for Australia and New Zealand combined.

Distribution of PID registrations. a Regional distribution of registered patients as a percentage of total patients (n = 1,209) in each state and territory of Australia, and in New Zealand. b Population adjusted study prevalence by region expressed as the number of patients per 100,000 population registered in each state and territory of Australia, and in New Zealand.). NSW = New South Wales, NT = Northern Territory, NZ = New Zealand, QLD = Queensland, SA = South Australia, TAS = Tasmania, VIC = Victoria, WA = Western Australia.
Underreporting is a key reason for patient registries to underestimate true disease prevalence [1]. We compared nationally acquired data on IVIg issued for PID (grams) with data on IVIg use from the register to approximate the rate of underreporting for the register (see “Methods”). Comparisons indicate that the PID register has acquired data on approximately 33.8–37.0% of the total target population of subjects affected by these conditions. Extrapolating these ranges to the entire register enabled an approximation of the adjusted prevalence of PID in Australia of 13.2–14.5 per 100,000 population (Table II). These figures may apply less well to PID syndromes that are not treated with IVIg. The reporting rates in South Australia (measured prevalence, 12.4; population, 1.56 million) may be nearing true population prevalence levels.
Clinical Manifestations of Primary Immunodeficiency Diseases
Clinical manifestations of disease (data available for 492 patients, 40.7% of total patients) were highly diverse (Table III). Bronchiectasis is a potentially preventable, high-morbidity, long-term complication of PID. Bronchiectasis was identified in 20% of respondents overall, and was more common in antibody deficiency disorders (23%) than other forms of PID (5%). Allergic manifestations of disease (13%), eczema (7%), autoimmune inflammatory disease (5%), cytopenias (4%), and granulomatous disease (2%) together combine to illustrate the relatively high frequency of immune dysregulation in PID and the complexity of clinical management. Low rates of tuberculosis (0.2%) are consistent with a stable low incidence of tuberculosis in the general population of Australia of 5.4 per 100,000 population [11].
Immunoglobulin Replacement Therapy
Immunoglobulin replacement therapy (IRT) data were recorded in 837 cases of a possible 1,098 (76.2%) cases (diagnoses of HAE, complement defects, and defects of phagocyte function were excluded on the basis that IRT is not indicated in these conditions). Of these 837 cases, 592 (70.7%) of patients were receiving IRT across 30 diagnoses, using a mean dose of 289.6 g (SD 155.2 g) per person per annum. The rates of intramuscular and subcutaneous delivery were low (3.4%). Data on both dose and weight was available in 254 cases. The average dose per kilogram body weight was 0.42 g/kg (SD 1.9 g/kg). Three conditions (CVID, IgG subclass deficiency, and agammaglobulinemia) dominated IRT for PID overall (Fig. 3).

Immunoglobulin replacement therapy: the ten most common indications for use (also includes remaining use of IRT not accounted for by the ten most common indication).
Immunoglobulin Replacement Therapy in Antibody Deficiency Syndromes Characterized by Normal Serum Total IgG
Immunoglobulin replacement therapy for antibody deficiency syndromes characterized by normal serum total IgG is controversial [8, 12]. Despite this, IRT was commonly used in the following two conditions: IgG subclass deficiency (65.0% of IgG subclass deficiency patients were receiving IRT) and specific antibody deficiency (89.3% of specific antibody deficiency patients were receiving IRT). These conditions also accounted for a large proportion of total IRT use (26.5% total IRT patients, Fig. 3). The mean dose of IRT for antibody deficiency syndromes with normal total serum IgG was 274.7 g (SD 114.3 g) per person per annum.
Discussion
Primary immunodeficiency diseases in Australia and New Zealand are prevalent, clinically diverse, geographically dispersed, and are characterized by high rates of potentially preventable morbidity and resource utilization. The ASCIA PID register provides a unique profile of PID enabled by relatively high rates of reporting (up to 12.4 per 100,000 in the state of South Australia), identification of 1,209 patients from a total population base of 24.7 million, and participation from 88 clinical centers. This report emphasizes the numerical importance of antibody deficiency disorders, including a large population of patients with antibody deficiency syndromes with normal serum IgG (30.1% of total patients) for whom clinical consensus is largely lacking [8, 13].
Although patient registries have an essential role in the mobilization of a public health response to PID, voluntary reporting imposes common limitations in the assessment of the overall population-based burden of disease [1]. In particular, data acquisition is subject to ascertainment biases (underreporting and recall biases) that are likely to favor patients with more severe disease or frequent attendance. Where possible the ASCIA PID register has been designed to mitigate these biases. First, the register was designed with web-based tools to provide ease of access to the central resources and documentation. Second, a network of PID officers was established across both countries to facilitate reporting. Third, register data acquired via voluntary reporting were compared with national Australian data on IVIg distribution, enabling estimates of underreporting. Fourth, ASCIA itself enabled the mobilization of the clinical immunology specialty toward data collection. Nevertheless, nonresponder biases may have important unmeasured effects. For example, this report may underestimate the significance of diseases commonly managed outside the specialty of clinical immunology such as congenital neutropenias in hematology, complex genetic phenotypes in pediatrics, or pathogen-specific immunodeficiencies [14] in the specialty of infectious diseases.
Prevalence Estimates
Population-based studies of PID prevalence in Australia and New Zealand are not available. Whereas the study prevalence of PID in the Australian component of the ASCIA PID register (5.6 per 100,000) is well below our adjusted prevalence estimate of 13.2–14.5 per 100,000 population (Table II), the measured South Australian prevalence of 12.4 per 100,000 exceeds other registries internationally: Norway 6.82 [15], Spain 5.07 [16], and Ireland 2.9 [17]. Intravenous immunoglobulin resource allocation for PID may prove to be a useful method of developing more accurate estimates of PID prevalence for those disorders for which IVIg is indicated. IgA deficiency is commonly reported to affect Caucasian populations at a rate of 1:700 [8], which could result in an Australasian population of more than 35,000 IgA-deficient subjects, the majority of whom would be asymptomatic. The ASCIA PID register did not actively recruit patients with asymptomatic IgA deficiency or other common variants such as mannose-binding lectin deficiency, which should be considered in the interpretation of the true population prevalence of PID.
Challenges for Service Delivery
-
1.
Geographic variation
Variations in regional reporting are most likely to be explained by local deficits in the clinical immunology workforce resulting in a lack of project participation or underdiagnosis. For example, low rates of reporting in the Northern Territory and Tasmania (Fig. 2) correlate with the absence of resident clinical immunologists [10]. Regional differences are less likely to be affected by true prevalence variations or migration as supported by the lack of variation in the profile of diagnoses by region. Even within reporting immunology centers, small clinical caseloads impose a challenge to service delivery and the mobilization of national programs in research, education, and clinical trials. Further studies would be required to determine whether patients managed outside the specialty of immunology, or those attending centers with small caseloads experience diagnostic delay or unsatisfactory clinical outcomes. Given that regional underreporting may link with access to local specialist services, the immunology workforce in key regions should be reviewed.
-
2.
Cellular and molecular diagnostics
The gap between scientific advances in PID and changes to clinical practice is particularly evident when considering molecular diagnosis, achieved in only 5.5% of cases. Molecular genotyping enables precise definition, early diagnosis, and improved long-term outcomes [19]. Clinical molecular genotyping is both complex and expensive [18] and for the majority of the most common diagnoses, the precise gene defects have not been established. These findings support the need for programs directed toward cellular and molecular discovery, mobilization of diagnostic services, and cost reduction enabled by economies of scale. Given the geographic dispersal of patients, such programs are likely to be beyond the means of individual departments and require strategic support from government and granting authorities. Australasia lacks the necessary coordinated public health focus on PID programs that are becoming established elsewhere [1, 2].
Potentially Preventable Complications of Disease
Chronic suppurative lung disease and bronchiectasis are potentially preventable complications of PID, especially in antibody deficiency syndromes [19]. The high prevalence of bronchiectasis in PID may be a product of delayed diagnosis [20], lack of multidisciplinary intervention [19], and inadequate immunoglobulin replacement [21]. The ASCIA PID register design may favor acquisition of patients with severe disease such as bronchiectasis. Despite this, rates of bronchiectasis of 20% overall, and 23% of patients with antibody deficiency syndromes raise a public health issue of adequacy of diagnostic awareness and intervention. These rates provide indicators for both international comparison and future analysis of local service delivery. Greater collaboration between the disciplines of clinical immunology, respiratory medicine, and primary care may be required to improve respiratory outcomes. Furthermore, the scope of clinical manifestations of disease in PID, including allergic and inflammatory disorders as well as the signature feature of frequent infections, mandates multidisciplinary specialist clinical support for patients.
Immunoglobulin Replacement Therapy
Immunoglobulin replacement therapy is the cornerstone of treatment for antibody deficiency disorders (reviewed in [12]). Therapeutic pooled immunoglobulin use is growing internationally with expanded indications in both immunodeficiency and immunomodulatory settings. Immunoglobulin replacement therapy in Australia and New Zealand is delivered largely by the intravenous as opposed to subcutaneous route, but may well change in keeping with trends elsewhere. Clinical trials of specialized subcutaneous Ig products may see an expansion of subcutaneous IRT use in the near future.
Understanding how IRT is used in PID is a matter of great importance. Whereas IRT use in agammaglobulemic and hypogammaglobulinemic disorders in the context of recurrent infection generally lacks controversy, treatment of antibody deficiency states with normal serum IgG are less substantiated [8, 12]. Nevertheless, IgG subclass deficiency and specific antibody deficiency were common indications for treatment with IRT in the register (Fig. 3). Many of these patients had reported clinical manifestations of disease consistent with a CVID spectrum disorder. Furthermore, studies of cohorts of adult patients with idiopathic bronchiectasis uncover frequent cases with incomplete defects of antibody production [22]. These findings of frequent reporting, burden of infectious complications, and extent of IRT utilization mandate further studies of the disease concepts underlying antibody deficiency states with normal serum IgG levels.
The Future
This report of current knowledge of PID in Australia and New Zealand highlights the need for a multifaceted, coordinated strategy for research and service delivery. Key areas of concern are population-based epidemiological studies, strategies to improve patient outcomes, access to molecular diagnostics, clinical immunology workforce review, and IRT governance. Further studies are required in antibody deficiency states with normal serum IgG levels.
Abbreviations
- ASCIA:
-
Australasian Society of Clinical Immunology and Allergy
- CVID:
-
common variable immunodeficiency
- IRT:
-
immunoglobulin replacement therapy
- IVIg:
-
intravenous immunoglobulin
- PID:
-
primary immunodeficiency disease
- SCID:
-
severe combined immunodeficiency
References
Lindegren ML, Kobrynski L, Rasmussen SA, Moore CA, Grosse SD, Vanderford ML, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. Morb Mortal Wkly Rep 2004;53:1–29.
European Commission. Consensus report and recommendations. in: Proceedings of the European Primary Immunodeficiencies Consensus Conference, Langen, Germany; 2006.
Notarangelo L, Casanova JL, Conley ME, Chapel H, Fischer A, Puck J, et al. Primary immunodeficiency diseases: an update. J Allergy Clin Immunol 2006;117:883–96.
Baumgart KW, Britton WJ, Kemp A, French M, Roberton D. The spectrum of primary immunodeficiency disorders in Australia. J Allergy Clin Immunol 1997;100:415–23.
Australian Bureau of Statistics. Australian Demographic Statistics: 3101.0; June 2006.
Statistics New Zealand. National Population Estimates; June 2006.
Aittoniemi J, Koskinen S, Laippala P, Laine S, Miettinen A. The significance of IgG subclass deficiency and mannan-binding lectin (MBL) for susceptibility to infection in apparently healthy adults with IgA deficiency. Clin Exp Immunol 1999;116:505–8.
Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005;94:S1–63.
Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol 2005;5:880–92.
Australasian Society of Clinical Immunology and Allergy. Clinical Immunology and Allergy Workforce Survey; 2007.
Roche P. Tuberculosis notifications in Australia, 2004. Commun Dis Intell 2006;30:93–101.
Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol 2006;117:S525–53.
Buckley RH. Immunoglobulin G subclass deficiency: fact or fancy? Curr Allergy Asthma Rep 2002;2:356–60.
Casanova JL, Fieschi C, Bustamante J, Reichenbach J, Remus N, von Bernuth H, et al. From idiopathic infectious diseases to novel primary immunodeficiencies. J Allergy Clin Immunol 2005;116:426–30.
Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol 2000;20: 477–85.
Llambi MJ, Etxagibel Galdos A, Matamoros N. The Spanish Registry of Primary Immunodeficiencies (REDIP). Allergol Immunopathol (Madr) 2001;29:122–5.
Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol 2005;25:73–7.
Costabile M, Quach A, Ferrante A. Molecular approaches in the diagnosis of primary immunodeficiency diseases. Hum Mutat 2006;27:1163–73.
Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. Q J Med 2002;95:655–62.
Seymour B, Miles J, Haeney M. Primary antibody deficiency and diagnostic delay. J Clin Pathol 2005;58:546–7.
De Gracia J, Vendrell M, Alvarez A, Pallisa E, Rodrigo MJ, de la Rosa D, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol 2004;4:745–53.
Vendrell M, de Gracia J, Rodrigo MJ, Cruz MJ, Alvarez A, Garcia M, et al. Antibody production deficiency with normal IgG levels in bronchiectasis of unknown aetiology. Chest 2005;127:197–204.
Acknowledgements
The ASCIA PID register receives unrestricted educational grants from CSL Bioplasma, ASCIA, and Octapharma Ltd. We thank the National Blood Authority of Australia for data on IVIg use and supply. We gratefully acknowledge all individuals who were involved in registering patients at their reporting centers. We also thank K Baumgart, L Bielby, W Britton, M Codarini, M Cook, E Elliot, M French, K Jayne, A Kakakios, A Kemp, R Loblay, K Maddock, M Schmidt, J Smith, A Turner, R Walls, and A Woodhouse for their invaluable contribution to this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article was written by the authors on behalf of the Australasian Society for Clinical Immunology and Allergy Primary Immunodeficiency Diseases Committee, Australia and New Zealand, which is also composed of E Benson, K Bleasel, W Britton, M Cook, M Empson, D Gillis, R Loh, J Peake, M Tang, J Sinclair, J Smart, M Wong, and J Ziegler.
Rights and permissions
About this article
Cite this article
Kirkpatrick, P., Riminton, S. Primary Immunodeficiency Diseases in Australia and New Zealand. J Clin Immunol 27, 517–524 (2007). https://doi.org/10.1007/s10875-007-9105-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-007-9105-z