
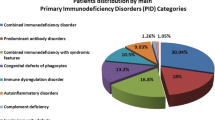
One hundred and twenty-four patients (from 120 families) diagnosed as primary immunodeficiency diseases were enrolled from five tertiary medical centers. The distribution by an update eight categories showed 45 patients (13 females/32 males; 36.3%) with “predominant antibody deficiencies,” 27 patients (6/21; 21.8%) with “T- and B-cell immunodeficiency,” 25 patients (9/16; 20.2%) with “congenital defects of phagocyte,” 25 patients (4/21; 20.2%) with “other well-defined immunodeficiency syndromes,” one boy (0.8%) with “disease in immune deregulation” (Chediak-Higashi syndrome) and another with “complement 3 deficiency.” None had “defects in innate immunity” or “auto inflammatory disorders.” Pseudomonas and Salmonella spp. were the two most identified microorganisms in septicemia (39.7%; 27/68 episodes). Twenty-three patients (18.5%) had mortality. Stem cell transplantation succeeded in 7 of 12 patients. In addition to nine patients with DiGerge syndrome recognized by FISH, direct sequencing identified 12 unique mutations from 20 families, reflecting distinct Taiwan geography, although a selection bias may exist.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Primary immunodeficiency diseases (PIDs) are a group of diseases that present as an unusual increased susceptibility to infections, characterized by 10 distinct warning signs (1). Since Bruton's first description of a patient with agammaglobulinemia and recurrent sinopulmonary infections in 1952 (2), about 100 different types of PIDs have been recognized (1, 3). For some patients, however, little distinct presentations and lack of convenient diagnostic approaches have resulted in inappropriate treatment for several years until critical health events and irreversible sequel were precipitated (3). Recently, advanced techniques of molecular diagnosis and exploring knowledge of immunology increase in the recognition rate of PIDs, subsequently revise an update categories (4).
Epidemiological studies have revealed wide geographical and racial variations in the incidence or prevalence and distribution patterns of PIDs. Most developed countries have their own clinical pictures and molecular basis of PIDs. However in Taiwan, we currently do not have sufficient comprehension in systemic approach to patients with PIDs. Extending our investigation from a single pediatric tertiary hospital to nationwide (5), the aim of this study was to speculate the distribution, infections, treatments and molecular analysis in Taiwanese patients with PIDs on the basis of an updated eight categories (4).
PATIENTS AND METHODS
Data Collection
Since July 1985 to June 2005, patients diagnosed as PIDs from a computer database search in Chang Gung Memorial Hospital (CG) were enrolled. Secondary immunodeficiencies were excluded as a previous study (5). Reported cases (6–15) and new patients in CG or referred from National Taiwan University Hospital (NT), Mackay Memorial Hospital (MK), Veterans Hospital (Ve), and Kaohsing University Hospital (Ks) were all collected. Patients with International Classification of Disease, Ninth Revision (ICD-9) number regarding PIDs were reclassified by an update eight categories, including “predominant antibody deficiencies,” “T- and B-cell immunodeficiency,” “congenital defects of phagocyte,” “other well-defined immunodeficiency syndromes,” “disease in immune deregulation,” “defects in innate immunity,” “auto inflammatory disorders,” or “complement deficiency.”
Analysis of Candidate Genes
After informed consent obtained from patients, 10–20 ml of venous blood was sent to our laboratory within 24–72 h. Extraction of RNA and synthesis of complementary DNA were performed as described previously (5). One or two pairs of oligonucleotide primers were selected for each gene to cover the entire coding region (5, 16, 17). The mutations identified from cDNA were confirmed by sequence analysis of genomic DNA. The individual exons, including the exon-intron boundaries, were amplified using designed primers (5, 16, 17). The 22q11.2 deletion responsible for DiGeorge syndrome (DGS) was detected by fluorescence in situ hybridization (FISH) (18). Expression of candidate molecules and/or proteins was evaluated using flow cytometry or immunostaining for anti-Btk antibody (mouse IgG1, provided by T. Futatani MD, and H.D. Ochs MD, University of Washington Medical Center, Seattle, WA), CD40L, IL-2 receptor common gamma chain (IL2RG or CD132), IL-12 receptor β1 (IL-12RB1), CD11b, CD18, human leukocyte antigen (HLA) class I (HLA-A, B, C) and class II (HLA-DR, DP, DQ; all purchased from Pharmingen, San Diego, CA), and Wiskott-Aldrich Syndrome Protein (WASP) antibodies (rabbit IgG1, a gift from Q. Zhu MD, University of Washington Medical Center, Seattle, WA) (5, 16–20).
RESULTS
Distribution in a Large Cohort of Patients with PIDs
The medical records of 124 patients from 120 unrelated families were collected from 5 tertiary medical centers. Fifty-three cases were from Chang Gung Memorial Hospital, a tertiary medical center responsible for 2,355,497 live births within the most recent 20 years (21), which corresponded to an estimated incidence of 2.2 per 100,000 live births. Together with patients from other hospitals, the distribution and clinical features of this cohort were summarized in Tables I and II. “Predominant antibody deficiencies” were the most common cases in 45 patients (36.3%). “T- and B-cell immunodeficiencies,” “congenital defects of phagocyte,” and “other well-defined immunodeficiency syndromes” were almost equal (approximate 20%) and the next common. “Immune deregulation” (Chediak-Higashi syndrome) and “complement 3 deficiency” were only found in one boy each (below 1%), respectively. None had “defects in innate immunity” or “auto inflammatory disorders.” Forty-two individuals (from 39 unrelated families) had a family history of PIDs. No antenatal diagnosis or consanguinity was traced.
Common Presentations, Infections and Mortality
One patient with Chediak-Higashi syndrome that was reclassified into “disease of immune deregulation” by an update categories, was conventionally classified into “congenital defects of phagocyte” (Table II) in this study. The age of onset ranged from the first day of life to 24-years-old, commencing below 3 years for 69.4% of all. Recurrent sinopulmonary infections (otitis media, sinusitis and/or pneumonia) were the most common presentations in all except “congenital defects of phagocyte” in this category severe skin infections and septicemia were more common. Splenomegaly and/or hepatomegaly in patients with “other well-defined immunodeficiency syndromes” were as common as recurrent sinopulmonary infections.
Fifty-two patients (33.9%) endured 68 episodes of identified septicemia, mainly Gram-negative bacteria in 46 episodes (Pseudomonas and Salmonella infections in 15 and 12; respectively as Table III).
The causes for mortality in 23 patients were infections (n=14; four associated with unsuccessful transplantation), malignancy (n=4; one related to EBV-induced lymphoproliferative disorders), complicated congenital heart diseases (n=3), veno-occlusive disease after transplantation (n=1), and disseminated intravascular coagulopathy (n=1) as Table IV.
Treatments
Regular IVIG infusions were given for patients with hypogammaglobulinemia and recurrent sinopulmonary infections (Table V) (22, 23). Short-term IVIG treatment (<4 doses) was administered in one individual with CVID and four with selective immunoglobulin deficiency (IgG2 subclass in 1, IgG2 and IgG4 subclass in 2, and IgG3 and IgG4 in 1) during the period of recurrent infections. Prophylactic antibiotics were prescribed in cases with T-cell defects, phagocyte defects and both predominant B-cell deficiencies and bronchiectasis. Granulocyte-colony stimulating factor (G-CSF) was administered to three patients with severe neutropenia and recurrent infections. Interferon-gamma (IFN-γ) treated two patients with IFN-γ-associated immunodeficiency when one had intractable salmonella infection and another had severe mycobacteria infections. IFN-γ also treated seven CGD patients to decrease the rate of severe infections, however, one of them died of Enterobacter cloacae sepsis while waiting for transplant donors. Stem-cell transplantation succeeded in seven cases (SCID [n=4], WAS [n=2], CHS [n=1]) but failed in five (SCID [n=2], WAS [n=2], LAD [n=1]) as Table VI. Engraftment succeeded in two SCID patients who received related HLA-matched bone borrow neither myeloablation nor GvHD prophylaxis, and another SCID patient received unrelated four-matched cord-blood transplantation at 5 months of age because of none matched bone marrow available from parents, siblings or volunteers, and achieved IVIG-independent immune reconstruction at 6 months post transplant.
Molecular Analysis
Molecular analysis was studied in 33 patients from 29 unrelated families (Table VII). We directly sequenced candidate genes in 24 patients and evaluated the expression level of candidate proteins in 22 patients, respectively as Table VII. As expected, the expressional level of translated proteins from causative genes were decreased or absent.
Twenty biologic parents received the carrier detection. All were carriers except for one with de novo mutated IL2RG (Trp 74 Gly).
DISCUSSION
The study from a large cohort of Taiwanese patients with PIDs shows that predominant antibody deficiencies (B cells) are the most common, similar to the worldwide reports (1). According to an update eight categories (4), the phenotypes and characterized laboratory findings are easily recognized in patients with SCID (12 cases) and HIGM (10 cases) [classified to “T- and B-cell immunodeficiencies”], WAS (10 cases) and DGS (9 cases) [“other well-defined immunodeficiency syndromes”], CGD (9 cases) and HIES (8 cases) [“congenital defects of phagocytes”]. These PIDs could be often diagnosed by well-experienced physicians referring simple laboratory tests. Thus, such three categories were quickly reminded and became the approximately second common cases. None patient with “defects in innate immunity” (for instant, anhidrotic ectodermal dysplasia with immunodeficiency [EDA-ID]) or “auto inflammatory disorders” (for example, familial Mediterranean fever or other period fever syndromes) may in part ascribe to racial variation, but the lower incidence rate (estimated at 2.2 per 100,000 live births), compared to 2.7 per 100,000 in Singapore (24) and 8.4 per 100,000 in Sweden (25), reflects an underestimation of the burden of patients with PIDs, especially for those genetically ill-defined patients with adult-onset cases of CVID (onset age over 24 years of age), a polysaccharide deficiency but normal immunoglobulins, and, extremely critical cases that did not survive before referral to medical centers. Moreover, patients with single-gene mutation constitute approximately 40.0% in other countries (1), but as high as 60.0 % in Taiwan (74/124 patients as Table I). The ratio also implies an underestimation in those patients with ill-defined genetic basis of PIDs.
In this cohort, clinical manifestations of recurrent sinopulmonary infections and septicemia are common in patients with PIDs. Similar infectious pathogens as other series, choosing empiric antibiotics to presumed pathogens of Strep. Pneumonia (in airway infections), pseudomonas and salmonella (in sepsis) are life saving until proven other else. Catalase-producing pathogens (e.g., Staph. aureus and Aspergillus spp) usually infect patients with phagocytic defects. Opportunistic infections (e.g., PCP and cytomegalovirus) often occur in patients with T-cell defects, containing “T- and B-cell” and “other well-defined immunodeficiency syndromes” by update categories (4). Compared to worldwide 1233 patients (63.0% of 1956 patients) with successful hematopoietic stem cell transplantation (HSCT) since 1968–1997 (26), only seven patients (58.3% of 12 patients) succeeded within the most recent 20 years. Thirteen cases deceased while waiting for suitable HSCT. Encouraging, a SCID patient with de novo IL2RG mutation received unrelated four-matched umbilical cord HSCT (UCSCT). One year later, he has 82% donor chimerism in the lymphocytes and recovery of T-cell function. UCSCT is an alternative source for patients with critical status of severe PIDs, including SCID, CGD, WAS, XLP, and DGS (27–32). Compared with bone marrow, the benefits of UCSCT in earlier recovery of immune function, lower GvHD risk, and lower viral transmission rate are proposed (33), but long-term prognosis will be not yet determined.
The fundamentally conservative nature of Taiwan culture released just 33 patients (from 29 unrelated families) for molecular analysis, revealing twelve unique mutations (14 patients from 12 families) from 20 identified mutations that are not located on hot spots (website://bioinf.uta.ti). The higher percentage of novel mutations (60.0%, 12/20) reflects distinct Taiwan geography.
For developing comprehensive molecular diagnosis in Taiwanese patients with PIDs, we continue elucidating genetic basis of CVID-the most popular disease in PIDs. CVID is a heterogeneous syndrome which “masks” or overlaps disorders in patients with mutations of the Btk, CD40L, SH2D1A/DHSP/SAP or ICOS genes (16, 17, 34). We analyzed these four possible candidate genes in 12 CVID patients (3/9 F/M). Subsequently, a mutated [Asp521Val] Btk gene in two cousins and [Lys96Stop] CD40L in a boy were identified in Table VII, initially diagnosed as CVID. None mutation was found in the SH2D1A/DHSP/SAP and ICOS genes. In most recent, taking emerging concept that the BAFF/Blys signaling (B-cell-activating factor of the tumor necrosis factor family) enhances B-cell survival, CD40L (T-cell) independent antibody isotype switching, and germinal center maintenance through three receptors mainly on the surface of B cell (35–37): BAFFR (BAFF receptor), TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor) and BCMA (B-cell maturation antigen). Such observation in gene knock-out mice led to the exploration of patients with mutations of BAFFR and TACI from the Caucasian CVID cohort by Grimbacher and Geha study groups (38–40). Meanwhile, mutations of CD19, caspase-8 and caspase-9 could have the CVID phenotype (41, 42). These new causative genes are the ongoing subjects to investigate CVID patients.
In conclusion, our experience reported here shows that predominate antibody deficiencies, found in many studies more 60% (1), is only 36.6%, more likely that it is the absence of adult-onset PIDs, especially for adult-onset CVID. Ethic factors may contribute to the higher unique mutations in the isolated Formosa Island, Taiwan. This review is to raise awareness in physicians rather than pediatricians, and keep tune for exploring knowledge to identify new candidate genes in PIDs. Clinically, high index of suspicion, well-control infection, regular IVIG and optimal HSCT will rescue more PIDs patients.
Abbreviations
- PIDs:
-
primary immunodeficiency diseases
- HIGM:
-
hyper IgM syndrome
- NEMO:
-
nuclear factor kB (NF-κB) essential modulator
- ICOS:
-
inducible co-stimulatory molecule
- ICOSL:
-
ICOS ligand
- CVID:
-
common variable immunodeficiency
- CD40L:
-
CD40 ligand
- AID:
-
activation-induced cytidine deaminase
- SAP:
-
signaling lymphocyte activation molecule-associated protein
- WASP:
-
Wiskott-Aldrich syndrome protein
- AT:
-
ataxia telangiectasia
- HIES:
-
hyper IgE syndrome
- LAD:
-
leukocyte adhesion disease
- CHS:
-
Chediak-Higashi syndrome
- SCID:
-
severe combined T- and B-cell immunodeficiency
- CGD:
-
chronic granulomatous disease
- GvHD:
-
graft vs. host disease
- PBMC:
-
peripheral blood mononuclear cells
- RT-PCR:
-
reverse transcriptase polymerase chain reaction
- FISH:
-
fluorescence in situ hybridization.
REFERENCES
Smith CIE, Ochs HD, Puck JM: Genetically determined immunodeficiency diseases: A perspective. In Primary Immunodeficiency Diseases, HD Ochs, CIE Smith, JM Puck (eds). New York, Oxford, 1999, pp 3–11
Bruton OC: Agammaglobulinemia. Pediatrics 9:772–778, 1952
Stiehm ER, Ochs HD, Winkelstein JA: Immunologic disorders: General considerations. In Immunodeficiency Disorders in Infants and Children, ER Stiehm, HD Ochs, JA Winkelstein (eds). Philadelphia, WB Saunders, 2004, pp 289–355
Notarangelo L, Casanova JL, Fischer A, Puck J, Rosen F, Seger R: Primary immunodeficiency diseases: An update. J Allergy Clin Immunol 114:677–687, 2004
Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ: Distribution and clinical aspects of primary immunodeficiencies in a Taiwan Pediatric tertiary hospital during a 20-year-period. J Clin Immunol 25:162–173, 2005
Yang YL, Lu MY, Jou ST, Lin KH, Lin DT: Hematopoietic stem cell transplantation in Taiwanese children with primary immunodeficiency. J Formos Med Assoc 104:101–106, 2005
Weng JD, Shyur SD: X-linked chronic granulomatous disease: Report of one case. Acta Paediatr Tw 45:163–167, 2004
Hung CH, Cheng SN, Hua YM, Wang CL, Chen YH, Yang KD: Leukocyte adesion deficiency disorder: Report of one case. Acta Paediatr Sin 40:128–131, 1999
Chien YH, Hwu WL, Ariga T, Chang KW, Yang YH, Lin KH, Chiang BL: Molecular diagnosis of Wiskott-Aldrich syndrome in Taiwan. J Microbiol Immunol Infect 37:276–281, 2004
Lin SC, Shyur SD, Ma YC, Huang LH, Lee WI: Hyper-IgM1 syndrome with interstitial pneumonia and diarrhea caused by coxsackievirus B4 in a 3-month-old infant. Ann Allergy Asthma Immunol 95:93–97, 2005
Ma YC, Lee WI, Lin SC, Huang LH, Wu JY: De novo mutation causing X-linked hyper-IgM syndrome: A family study in Taiwan. Asian Pac J Allergy Immunol 23:53–59, 2005
Lin SJ, Huang YF, Chen JY, Heyworth PG, Noack D, Wang JY, Lin CY, Chiang BL, Yang CM, Liu CC, Shieh CC: Molecular quality control machinery contributes to the leukocyte NADPH oxidase deficiency in chronic granulomatous disease. Biochim Biophys Acta 1586:275–286, 2002
Wang LH, Tsai MJ, Huang MT, Lin SC, Chiang BL: Autoimmune manifestations in patients with primary immunodeficiency. Acta Paediatr Tw 40:243–249, 1999
Wang LJ, Yang YH, Lin YT, Chiang BL: Immunological and clinical features of pediatric patients with primaty hypogammaglobulinemia in Taiwan. Asian Pac J Allergy Immunol 22:25–31, 2004
Hung CH, Hua YM, Huang CF, Luo WT, Yang KD, Chu ML, Wang CC: Chronic granulomatous disease: A case report. J Formos Med Assoc 100:281–284, 2001
Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD: Mutation analysis in a large cohort patients with hyper IgM syndrome. Blood 105:1881–1890, 2005
Lee WI, Zhu Q, Gambineri E, Jin Y, Welcher AA, Ochs HD: Inducible CO-Stimulator molecule (ICOS), a candidate gene for defective isotype switching, is normal in patients with Hyper IgM syndrome of unknown molecular diagnosis. J Allergy Clin Immunol 112:958–964, 2003
Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS: Prevalence of 22q11 microdelletions in DiGeorge and velocardiofacial syndromes: Implications for genetic counseling and prenatal diagnosis. J Med Genet 30:813–817, 1993
Sakai T, Matsuoka M, Aoki M, Nosaka K, Mitsuya H: Missense mutation of the interleukin-12 receptor β1 chain-encoding gene is associated with impaired immunity against mycobacterium avium complex infection. Blood 97:2688–2694, 2001
Monis-Teisserenc HT, Gadola SD, Cella M: Association of a syndrome rembling Wegener's granulomatosis with low surface expression of HLA-class-I molecules. Lancet 354:1598–1603, 1999
Report on Registration of Births and Deaths, 2004. Registry of Births and Deaths, National Registration Department, Taiwan
Chapel HM, Webster ADB: Assessment of the immune system. In Primary Immunodeficiency Diseases, HD Ochs, CIE Smith, JM Puck (eds). New York, Oxford, 1999, pp 419–431
Quartier P, Debre M, De Blic J, de Sauverzac R, Sayegh N, Jabado N, Haddad E, Blanche S, Casanova JL, Smith CI, Le Deist F, de Saint Basile G, Fischer A: Early and prolonged intravenous immunoglobulin replacement therapy in childhood aggammaglobulinemia: a retrospetic survey of 31 patients. J Pediatr 134:589–596, 1999
Lim DL, Thong BY, Ho SY, Shek LP, Lou J, Leong KP, Chng HH, Lee BW: Primary immunodeficiency diseases in Singapore-the last 11 years. Singapore Med J 44:579–586, 2003
Fasth A: Primary immunodeficiency disorders in Sweden: cases among children. 1974–1979. J Clin Immunol 2:86–92, 1982
Buckley RH, Fischer A: Bone marrow transplantation for primary immunodeficiency diseases. In Primary Immunodeficiency Diseases, HD Ochs, CIE Smith, JM Puck (eds). New York, Oxford, 1999, pp 459–475
Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, Myers LA, Ward FE: Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 340:508–516, 1999
Knutsen AP, Wall DA: Kinetics of T-cell development of umbilical cord blood transplantation in severe T-cell immunodeficiency disorders. J Allergy Clin Immunol 103:823–832, 1999
Bhattacharya A, Slatter M, Curtis A, Chapman CE, Barge D, Jackson A, Flood TJ, Abinun M, Cant AJ, Gennery AR: Successful umbilicord blood stem cell transplantation for chronic granulomatous disease. Bone Marrow Transplant 31:403–405, 2003
Ozsahin H, Le Deist F, Benkerrou M, Cavazzana-Calvo M, Gomez L, Griscelli C, Blanche S, Fischer A: Bone marrow transplantation in 26 patients with Wiskott-Aldrich syndrome from a single center. J Pediatr 129:238–244, 1996
Ziegner UH, Ochs HD, Schanen C, Feig SA, Seyama K, Futatani T, Gross T, Wakim M, Roberts RL, Rawlings DJ, Dovat S, Fraser JK, Stiehm ER: Unrelated umbilical cord stem cell transplantation for X-linked immunodeficiencies. J Pediatr 138:570–573, 2001
Ohtsuka Y, Shimizu T, Nishizawa K, Ohtaki R, Someya T, Noguchi A, Shimura N, Kim H, Sugimoto H, Fujita H, Morio T, Yamashiro Y: Successful engraftment and decrease of cytomagalovirus load after cord blood stem cell transplantation in a patient with DiGeorge syndrome. Eur J Pediatr 163:747–748, 2004
Thomson BG, Robertson KA, Gowan D, Heilman D, Broxmeyer HE, Emanuel D, Kotylo P, Brahmi Z, Smith FO: Analysis of engraftment, graft-versus-host disease, and immune recovery following unrelated donor cord blood transplantation. Blood 96:2703–2711, 2000
Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, Eibel H, Fischer B, Schaffer AA, Mages HW, Kroczek RA, Peter HH: Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol 4:261–268, 2003
Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, Tumas D, Grewal IS, Dixit VM: Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol 2:638–643, 2001
Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C: BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 293:2108–2111, 2001
Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, Bram RJ, Jabara H, Geha RS: TACI and BAFF-R mediate isotype switching in B cells. J Exp Med 201:35–39, 2005
Warnatz K, Salzer U, Gutenberger S, Schlesier M, Grimbacher B, Peter HH, Eibel H: Finally found: Human BAFF-R deficiency causes hypogammaglobulinemia. Clin Immunol Suppl:S20, 2005 [abstract]
Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, Peter HH, Rockstroh JK, Schneider P, Schaffer AA, Hammarstrom L, Grimbacher B: Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet 37:820–828, 2005
Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, Geha RS: TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet 37:829–834, 2005
Castano D, Patino PJ, Woellner C, Salzer U, Grimbacher B, Montoya CJ, Orrego JC, Rugeles C, Franco JL: Novel humoral immunodeficiency in humanns associated with deleterious homozygous mutation in CD19. Clin Immunol Supp1:S29, 2005 [abstract]
Chiocchetti A, Mesturini R, Bensi T, Biava A, Ferretti M, Santoro C, Pignata C, Rieux-Laucat F, Dianzani I, Ramenghi U, Notarangelo LD, Dianzani U: Hypogammaglobulinemia and lymphoproliferation in two patients with heterozygous deleterious mutation of the Caspase-9 gene. Clin Immunol Supp1:S241, 2005 [abstract]
ACKNOWLEDGMENTS
The authors would like to thank Bor-Luen Chiang MD, PhD in National Taiwan University hospital (NT), Shyh-Dar Shyur MD in Mackay memorial hospital (MK), Wen-Jue Soong MD in Veterans hospital (Ve), and Shyh-Shin Chiou MD, PhD in Kaohsing University hospital (Ks) for the referrals, all participating patients and families for their kind cooperation. We also wish to express our gratitude to Mai-Tzu Chen, Hsiu-Li Chou and Hsiu-Shan Hsiao for their technical assistance. This study was supported in part by Chang-Gung Medical Research Progress grants (CMRPG 32069 awarded to W.I. Lee and 32003 to J.L. Huang) and National Science Council grants (NMRPG 3131 and C 93-2314-B-182A-111 to W.I. Lee and 3116 to J.L. Huang).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, WI., Jaing, TH., Hsieh, MY. et al. Distribution, Infections, Treatments and Molecular Analysis in a Large Cohort of Patients with Primary Immunodeficiency Diseases (PIDs) in Taiwan. J Clin Immunol 26, 274–283 (2006). https://doi.org/10.1007/s10875-006-9013-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-006-9013-7