
Abstract
Introduction
Primary immunodeficiency disorders (PIDs) are heterogeneous disorders that mainly present with severe, persistent, unusual, or recurrent infections in childhood. Reports from different parts of the world indicate a difference between Western and Eastern populations.
Aim
The aim of this study was to report on the different patterns of PIDs and identify subgroup characteristics in a highly consanguineous population in Egypt.
Methods
We performed a retrospective chart review for children below 18 years diagnosed with PID at Cairo University Pediatric Hospital from 2010 to 2014.
Results
Four hundred seventy-six children were diagnosed with PID disorders. Major categories included combined immunodeficiency disorders, which constituted a large proportion (30 %) of cases, along with predominantly antibody disorders (18 %) followed by syndromic combined disorders (16.8 %), phagocytic disorders (13.2 %), immune dysregulation disorders (10.5 %), and autoinflammatory disorders (9 %).
Conclusion
PIDs have different patterns within inbred populations with high consanguinity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiency disorders (PIDs) constitute several hundred disease entities. Their main presentation is with severe, persistent, unusual, or recurrent infections. Other presentations may include autoimmunity, lymphoproliferation, and/or malignancy. PIDs are remarkably underreported in developing countries [1] because of several factors like attribution of infections to malnutrition and difficulties with diagnosis in limited resource settings with scarcity of needed investigations. Interestingly, PIDs in areas with high rates of consanguinity have demonstrated a peculiar prevalence of more severe forms of diseases and a different overall distribution [1, 2].
Egypt’s population has been estimated as 90 million in 2015. With an estimated birth rate of 23.35 births/1000, children constitute about 32 % of the population. Consanguinity, a deeply rooted cultural trend in Egypt, ranges from 35.3 to 60 % with higher rates in rural areas and causes a relative abundance in autosomal recessive disorders [3, 4].
The high rates of consanguinity and inbreeding result in high rates of autosomal recessive inherited disorders as documented in other diseases studied in consanguineous populations [5, 6].
The study aims to report on the spectrum and different patterns of PID in Egyptian children within a 5-year interval of PID search at a tertiary referral center located in Cairo and to identify subgroup characteristics in a highly consanguineous population.
Methods
The study was set at the Primary Immunodeficiency Clinic at Cairo University Pediatric Hospital and Immunology Laboratory, Department of Clinical and Chemical Pathology, Cairo University. The clinic receives referrals from all over the country and neighboring countries. The study was designed as a retrospective chart review through 5 years from 2010 to 2014.
Children were referred for assessment from outpatient clinics and inpatient wards if they had warning signs suggestive of PID (severe, unusual, persistent, or recurrent infections) or specific phenotypes such as autoimmunity/lymphoproliferation/allergy suggestive of a certain immunodeficiency disorder, e.g., autoimmune lymphoproliferative syndrome (ALPS) (presenting with lymphoproliferation and autoimmune cytopenias) or syndromic features (hypocalcemia with congenital heart disease suggestive of DiGeorge syndrome). Cases were subsequently tested based on clinical suspicion of one of the PID categories. Diagnosis was established according to the criteria set by the European Society of Immunodeficiency Disorders (ESID) and classified according to the International Union of Immunological Societies [7]. Cases excluded from the study were children proved to have other diagnoses responsible for their symptoms such as cystic fibrosis, anatomical defects, etc. The study protocol was approved by the Institutional Review Board (IRB). Children were examined by pediatricians who took a complete history and performed a clinical examination.
Laboratory Assessment
Complete blood counts and immunophenotyping of peripheral blood lymphocytes was done by flow cytometry. Lymphocytes were characterized by a panel of monoclonal antibodies: anti-CD3 as pan-T marker, anti-CD4 for T helper lymphocytes, anti-CD8 for cytotoxic T cells, anti-CD19 or anti-CD20 as pan-B markers, anti-CD16, and anti-CD56 for natural killer cells. Specific memory/naive markers as CD27, IgD, CD45RA, and CD45RO were used for determination of memory versus naive B and T cells when indicated. CD18/CD11 and Tregs were assayed in certain cases. Dihydrorhodamine assay was done in case of suspicion of phagocytic defects with analysis of specific deficient NADPH components for chronic granulomatous disease (CGD) patients by flow cytometry. Further tests were tailored according to the patient’s presentation, e.g., TCRαβ for query ALPS patients, WASP protein for suspected Wiskott-Aldrich patients, and DOCK8 in hyperimmunoglobulin E syndrome (HIES). Serum immunoglobulin levels were determined by nephelometry. Genetic analysis was done for some cases when available.
Statistics
Descriptive statistics were presented with mean, +/− standard deviation (SD), or median with interquartile range for continuous variable and proportion of categorical variables.
Results
During the period from 2010 to 2014, a total of 5000 children were screened for PID. Out of those, 476 were identified with confirmed PID disorders. One hundred twenty children were diagnosed with probable PID (not matching specific disease criteria or not having enough evidence to support the diagnosis yet presenting with clinical manifestations highly suggestive of PID). The patients were diagnosed with 30 different primary immunodeficiency disorders which belonged to eight different categories (Table 1).
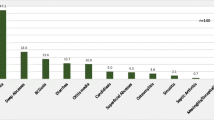
T cell/combined disorders constituted 143 (30 %) and were the most common disorders, followed by predominantly antibody disorders with 86 (18 %), syndromic combined disorders with 80 (16.8 %), phagocytic defects of number or function with 63 (13.2 %), immune dysregulation with 50 (10.5 %), autoinflammatory disorders with 43 (9 %), complement deficiency with 6 (1.3 %), and defects of innate immunity with 5 (1 %) (Fig. 1).

Primary immunodeficiency disorder distribution by category
The percentage distribution of PID categories in similar countries with high rates of consanguinity is demonstrated in Fig. 2 although each country’s cohort is different regarding intervals of case recruitment and sample size.

Percentage distribution of primary immunodeficiency disorders by different categories in Middle Eastern countries with high consanguinity compared to the European Society of Immunodeficiency Disorders Society’s (ESID) registry
Among the 140 cases diagnosed with severe combined immunodeficiency (SCID)/Omenn syndrome, sufficient data for categorization was available for only 91 cases over the past 3 years. Of those, 46 (50.5 %) cases were diagnosed with T-B-SCID, 35 (38.5 %) with T-B + SCID, and 10 were diagnosed with Omenn syndrome (11 %). RAG mutations were identified in four out of 10 children presenting with T-B-SCID/Omenn phenotype. Interestingly, one of the patients had a novel nonsense mutation (Q812X) detected in a homozygous form despite being born to nonconsanguineous parents [8]. Prenatal diagnosis was carried out for three pregnant mothers with previously affected SCID or Omenn syndrome infants having a homozygous RAG1 mutation. Two fetuses were diseased while the third had the wild-type gene.
T cell receptor excision circles (TRECs) assays were used for targeted screening in high-risk infants with a positive yield in 18.4 % (23/125 neonates) of studied cases diagnosed with SCID. Two SCID patients were successfully transplanted.
Among the categories of combined immunodeficiency with syndromic features, hyperimmunoglobulin E syndrome (HIES) was the most common diagnosis with 35 cases diagnosed based on their clinical features and elevated immunoglobulin E (IgE) level, which ranged between 2000 and 50,000 IU/ml. Out of 20 cases screened for genetic mutations, there were 12 DOCK8 deficiency cases, one case of STAT3 deficiency, and a group of children with typical clinical presentations, laboratory investigations, and unidentified genetic mutations. Thirty-two patients were diagnosed with ataxia telangiectasia; three of them developed malignancy in the form of acute lymphoblastic leukemia and Hodgkin and non-Hodgkin lymphomas.
Thirty-six patients were diagnosed with less than 2 % CD19 levels. Based on family history with exclusive maternal cousins’ and/or uncles’ affection, a presumptive diagnosis of X-linked agammaglobulinemia was made in 14 cases, and the rest were suggestive of autosomal recessive agammaglobulinemia.
Fifty patients were diagnosed with different immune dysregulation disorders including hypopigmentation disorders, hemophagocytic lymphohistiocytosis (HLH), autoimmune lymphoproliferative disease (ALPS), and immune dysregulation, polyendocrinopathy, enteropathy, X-linked immune deficiency (IPEX)/IPEX-like syndromes.
Chronic granulomatous disease (CGD) was the most commonly diagnosed disorder in the phagocytic disorder category. The group included 45 children, and data was retrievable for 31 children from 28 different kindred. Twenty-seven children were proved to have autosomal recessive CGD with the most common defective protein was p22-phox in 14 patients, followed by p47-phox in 11 patients, and p67-phox in 2 patients. Four children presented as X-linked CGD based on maternal bimodal dihydrorhodamine assay patterns and exclusive male affection.
Familial Mediterranean fever (FMF) was one of the most commonly diagnosed disorders, with 41 cases diagnosed based on mutations detected in the Mediterranean fever (MEFV) gene.
Clinical Presentation
Recurrent infections were the main presenting symptoms in 90 % of our cases, with pneumonia topping the list followed by chronic diarrhea, skin abscesses, and chronic otitis media. Family history of similar cases was positive in 80 %. A minority of cases presented with other manifestations, e.g., autoimmunity, malignancy, and/or lymphoproliferation. In the category predominantly antibody deficiency disorders, presentations extended to include recurrent sinopulmonary infections, bronchiectasis, cor pulmonale, and stunted growth. In hyperimmunoglobulin M, patients also suffered from sclerosing cholangitis, autoimmune cytopenias, and cryptosporidial diarrhea. In the CGD group, BCGosis following compulsory BCG vaccination was recorded in three patients. Fungal infections among those patients were very vigorous and frequently involved the central nervous system.
Age of Onset, Age of Diagnosis, and Diagnosis Delay
The age of onset of symptoms ranged between 0 and 9 years with mean of 1.15 years for all PID categories. While the age of diagnosis of the patients ranged between 0.08 and 17 years with mean of 3.25 years. Majority of the patients (83 %) were diagnosed during childhood (<14 years). Prenatal diagnosis confirmed the disease in two fetuses, and mothers decided to terminate the pregnancy. The overall mean delay in diagnosis was 1.67 years.
Genetic Analysis
Genetic analysis was not always available. In 106 patients (22.26 %), the genetic defect was identified. The identified mutated genes among SCID were RAG1&2 (6 patients), artemis (1 patient), and purine nucleoside phosphorylase deficiency (2 patients). Among the CGD group, NCF1 (8 patients), CYBA (14 patients), NCF2 (2 patients), and CYBB (4 patients). In HIES, mutations were detected in dedicator of cytokinesis 8/DOCK8 (12 patients), and STAT3 (1 patient). In all FMF patients, mutations were detected in the MEFV gene. Mutations were also detected in IL-10 RA, IL10RB, STAT1 (2 patients), CD40 (2 patients), IL-12RA (1 patient), IL-12RB (1 patient), and FAS (1 patient); there were 2 patients with ORAI, 1 patient with TCN2, and 1 patient with IFNGR1 gene mutation. One patient was diagnosed with NEMO deficiency (nuclear factor kappa B essential modulator).
Atypical Cases
We had cases presenting with multiple mutations and a mixed phenotype:
-
Case 1
A 9-year-old female presenting with pneumonia, diarrhea, draining ears, cholangitic microabscesses, portal venous thrombosis, oral candidiasis, and eczema was diagnosed with a homozygous deletion of exons 1–44 in DOCK 8 and a homozygous missense variant in exon 13 of CARD9.
-
Case 2
A male patient with a history of two siblings’ deaths by classical T-B-SCID. Sequencing revealed two homozygous missense mutations in RAG2 (p.T2151) and (p.R229Q). Both parents and grandmothers (sisters) carried the two mutations in a heterozygous form while the grandfather was normal, indicating both mutations to be on the same allele.
-
Case 3
A 5-year-old boy with pneumonia, sinusitis, otitis, eczema, diarrhea, recurrent warts, and multiple fractures was found to have a homozygous mutation in DOCK8 and two heterozygous mutations in the AIRE gene.
Discussion
This study reports on a different distribution of PID disorders in a consanguineous population with a prevalence higher than estimated and with low awareness among the population as well as healthcare providers.
The rate of consanguinity was obviously very high in all PID categories reported in our population. In combined immunodeficiency disorders, the consanguinity rate was almost 79.7 %, a rate that is very close to that of Saudi Arabia (82 %) but higher than that of Turkey (62 %), Tunisia (56 %), and Morocco (44.8 %) but less than that of Kuwait (100 %) [9–13]. While we reported a consanguinity rate of 85.7 % in our HIES patients, lower rates were reported in nearby countries; 77 % in Saudi Arabia, 60 % in Turkey, 39 % in Kuwait, and 37.5 % in Morocco [9, 12–14]. For antibody deficiency disorders, the consanguinity rate in our patients was 68.6 %, a rate close to that of Saudi Arabia (74 %) and Kuwait (70 %) but higher than that of Morocco (40.6 %) and Turkey (9.7 %) [9, 12, 13, 15]. Regarding patients with immune dysregulation, the consanguinity was 86 % with a rate close to that of Saudi Arabia (83 %) and Turkey (80 %) [9, 15], but higher than that of Morocco (44.4 %) and lower than that of Kuwait (100 %) [12, 13]. In CGD, the present study reported a consanguinity rate of 83 %, similar to Kuwait’s but higher than Saudi Arabia’s (74 %), Turkey’s (61.7 %), and Morocco’s (54.5 %) [9, 13, 16], while in LAD patients, the consanguinity rate (90 %) was higher than what was reported in other studies (Saudi Arabia [85 %], Morocco [38.5 %], Turkey [47.9 %]) [9, 12, 15]. With regard to autoinflammatory disorders, the consanguinity was observed in 65 % of the cases, a much higher rate than Turkey’s 12 % and Morocco’s 16.7 % while this category was not reported in Saudi Arabia and Kuwait [9, 12, 13, 15].
T cell/combined immunodeficiencies were reported as the most common class (30 % combined disorders and 46.8 % when combined with syndromic disorders) compared to the other categories as reported in similar populations like Tunisia (28.5 %), Saudi Arabia (59.7 %), and in a collective comparative study of the North African region with prevalence in Egypt, Tunisia, and Morocco, with 41, 27, and 19 % of cases, respectively [1, 9, 11].
Other countries demonstrate a high prevalence of combined disorders but not as the most common category as in Kuwait, where they came at 21 %, second to antibody disorders, and Oman, with a predominance of phagocytic disorder as the most common PID category [13, 17].
A previous study conducted in Egypt reported a total prevalence of combined T and B cell immunodeficiencies as (29.7 %) of their cohort of PID patients with a consanguinity rate of 62.5 % [18].
For SCID cases, identification of the genetic defect encourages proper counseling and has enabled parents to make informed decisions in their subsequent pregnancies. The study revealed SCID constituted a major fraction of PID disorders among our studied patients. The mean age at diagnosis was 8.9 months, which is quite late unfortunately. A study of an Egyptian cohort of PID patients also documented a high incidence of SCID with a relatively late age at diagnosis and a diagnostic lag of several months [18].
A study comparing SCID patients diagnosed in Kuwait and USA revealed a different age of onset and different molecular causes, which highlights the importance of newborn screening even if targeted among high-risk families [19].
Regarding North Africa, Morocco, as well as Tunisia, data showed that among T-B+ SCIDs, only a minority have a defective expression of the gamma chain (X-linked form), while autosomal recessive forms as interleukin 7 receptor alpha chain (IL7Rα) expression defect and janus kinase 3 (Jak-3) deficiencies were observed in a score of patients [1]. RAG mutations were confirmed to be not uncommon in the Egyptian SCID population as previously reported by Meshaal et al. [8].
Antibody deficiency disorders reported as the most common category of PIDs observed in European and Latin American registries were found to be lower in North African populations studied [1]. This was suggested by Barbouche et al., where agammaglobulinemia cases with absence of circulating B lymphocytes diagnosed in the three North African series were often highly suggestive of autosomal recessive mode of inheritance and confirmed in this study with a low incidence of X-linked agammaglobulinemia in comparison to other disorders [1].
In CGD, the most common defective proteins were p22-phox, then p47-phox, followed by gp91-phox, and finally p67-phox, indicating a predominance of the autosomal recessive mode of inheritance compared to the X-linked mode. These findings are in agreement with the study of CGD pattern in Turkish families by Köker and colleagues, which also demonstrated predominance of AR forms in a consanguineous population [16]. Other studies of the Israeli and Indian populations revealed the autosomal recessive patterns were 63 and 58 % of CGD population, respectively, also because of the high consanguinity rates [20, 21]. Western countries, on the other hand, show a predominance of X-linked CGD as the ESID registry reports an approximate two-thirds of CGD cases are X-linked forms [22, 23].
The clustering of specific protein deficiencies in particular geographical areas of our country probably points to a founder gene effect.
Cases of HIES screened for genetic mutations revealed a strong predominance of DOCK8 deficiency (AR form) as opposed to STAT3 mutations (AD form) in our patients. Interestingly, a group of typical HIES patients were negative for mutational testing by next-generation sequencing, suggesting an undiagnosed cause that might prevail in the community.
A study of the MEFV mutations in Egyptian patients with FMF revealed the most frequent gene mutations in the studied group were V726A, M694V, M680I, E148Q, and M694I at 41.2 %, 32.4 %, 29.4 %, 25 %, and 20.6 %, respectively. At least one of these main five founder mutations was present in 97.1 % of patients [24].
As for MSMD, a new IFNGR1 mutation underlying IFN-γR1 deficiency was detected among Egyptian children [25]. Similar cases were also reported frequently in a Tunisian series with IL12B and IL12Rβ1 gene defects and founder effects have been observed with interleukin 12 B (IL12B) gene mutation [26].
Interestingly, some of the cases had several coexisting disease-causing mutations and presented with a mixed phenotype.
In conclusion, the exact prevalence and/or incidence of PID disorders in Egypt remain a puzzling question in the absence of a national registry. This data set may act as a foundation for setting up a national record of PID disorders in Egypt.
The study demonstrates that in highly consanguineous populations, autosomal recessive forms of PID disorders prevail and may also have peculiar characteristics pertaining to presentations and underlying genetic mutations. Such populations constitute a rich high-potential area for the discovery of new genetic causes in the field of immune deficiency disorder research. The study also establishes that collaboration and networking allows a state-of-the-art approach to the genetic and functional characterization of genetic defects in families with primary immunodeficiency despite limited resources.
References
Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. 2011;1238:42–52.
Al-Herz W, Aldhekri H, Barbouche MR, Rezaaei N. Consanguinity and primary immunodeficiencies. Hum Hered. 2014;77(1–4):138–43.
Shawky RM, ElAwady M, Elsayed S, Hamadan G. Consanguinous matings among Egyptian population. Egypt J Med Hum Genet. 2012;12(2):157–63.
Temtamy S, Aglan M. Consanguinity and genetic disorders in Egypt. Middle East J Med Genet. 2011;1(1):12–7.
Anwar WA, Khayatti M, Hemminki K. Consanguinity and genetic diseases in North Africa and immigrants to Europe. Eur J Public Health. 2014;24 Suppl 1:57–63.
Bashamboo A, McElreavey K. Consanguinity and disorders of sex development. Hum Hered. 2014;77(1–4):108–17.
Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726.
Meshaal S., El Hawary R., Elsharkawy M., Mousa R., Farid R., Abd. Elaziz D, Alkady R, Galal N., Massaad M, Boutros J., El Marsafy A. Mutations in recombination activating gene 1 and 2 in patients with severe combined immunodeficiency disorders in Egypt. Clin Immunol. 2015; 158: 167–172
Al-Saud B, Al-Mousa H, Al Gazlan S, Al-Ghonaium A, Arnaout R, Al-Seraihy A, et al. Primary immunodeficiency diseases in Saudi Arabia: a tertiary care hospital experience over a period of three years (2010–2013). J Clin Immunol. 2015;35(7):651–60.
Azarsiz E, Gulez N, Karaca NE, Aksu G, Kutukculer N. Consanguinity rate and delay in diagnosis inTurkish patients with combined immunodeficiencies: a single center study. J Clin Immunol. 2011;31:106–11.
Mellouli F, Mustapha IB, Khaled MB, Besbes H, Ouederni M, Mekki N, et al. Report of the Tunisian Registry of Primary Immunodeficiencies: 25-Years of Experience (1988–2012). J Clin Immunol. 2015;35(8):745–53.
Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, et al. First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998–2012). J Clin Immunol. 2014;34:459–68.
Al –Herz W. Primary immunodeficiency disorders in Kuwait: first report from Kuwait National Primary Immunodeficiency Registry. J Clin Immunol. 2008;28(2):186–93.
Al Khatib S, Keles S, Garcia-Lloret M, Karakoc-Aydiner E, Reisli I, Artac H, et al. Defects along the Th17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. J Allergy Clin Immunol. 2009;124(2):342–8.
Kilic S, Ozel M, Hafizoglu D, Karaca NE, Asku G, Kutukculer N. The prevalence and patient characteristics of primary immunodeficiency diseases in Turkey—two centers study. J Clin Immunol. 2013;33:74–83.
Köker MY, Camcıoğlu Y, van Leeuwen K, Kılıç SŞ, Barlan I, Yılmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132(5):1156–63.
Al-Tamemi S, Elnour I, Dennison D. Primary immunodeficiency diseases in Oman: five years’ experience at Sultan Qaboos University Hospital. World Allergy Organ J. 2012;5(5):52–6.
Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol. 2009;29(3):343–51.
Al-Herz W, Notarangelo LD, Sadek A, Buckley R, USIDNET Consortium. Combined Immunodeficiency in the United States and Kuwait: comparison of patients’characteristics and molecular diagnosis. Clin Immunol. 2015;161(2):170–3.
Wolach B, Gavrieli R, de Boer M, Gottesman G, Ben-Ari J, Rottem M, et al. Chronic granulomatous disease in Israel: clinical, functional and molecular studies of 38 patients. Clin Immunol. 2008;129:103–14.
Rawat A, Singh S, Suri D, Gupta A, Saikia B, Minz RW, et al. Chronic granulomatous disease: Two decades of experience from a tertiary care center in North West India. J Clin Immunol. 2014;34(1):58–67.
Winkelstein JA, Marino MC, Johnston RB, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79:155–69.
Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11.
El-Garf A, Salah S, Iskander I, Salah H, Amin SN. MEFV mutations in Egyptian patients suffering from familial Mediterranean fever analysis of 12 gene mutations. Rheumatol Int. 2010;30(10):1293–8.
Galal N, Boutros J, Marsafy A, Kong XF, Feinberg J, Casanova JL, et al. Mendelian susceptibility to mycobacterial disease in Egyptian children. Mediterr J Hematol Infect Dis. 2012;4(1), e2012033.
Elloumi-Zghal H, Barbouche MR, Chemli J, Bejaoui M, Harbi A, Snoussi N, et al. Clinical and genetic heterogeneity of inherited autosomal recessive susceptibility to disseminated Mycobacterium bovis Bacille Calmette-Guérin infection. J Infect Dis. 2002;185:1468–75.
Acknowledgments
The authors wish to acknowledge the following scientists for their valuable support and provision of expertise to aid the diagnosis of our children:
M. Baker, J. Bustamanate, J.L. Casanova, T. Freiberger, R. Geha, B. Grimbacher, J. Litzman, M. Masaad, A. Villa
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Galal, N., Meshaal, S., Elhawary, R. et al. Patterns of Primary Immunodeficiency Disorders Among a Highly Consanguineous Population: Cairo University Pediatric Hospital’s 5-Year Experience. J Clin Immunol 36, 649–655 (2016). https://doi.org/10.1007/s10875-016-0314-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-016-0314-1