
Abstract
Obtaining site-specific assignments for the NMR spectra of proteins in the solid state is a significant bottleneck in deciphering their biophysics. This is primarily due to the time-intensive nature of the experiments. Additionally, the low resolution in the \(^{1}{\text {H}}\)-dimension requires multiple complementary experiments to be recorded to lift degeneracies in assignments. We present here an approach, gleaned from the techniques used in multiple-acquisition experiments, which allows the recording of forward and backward residue-linking experiments in a single experimental block. Spectra from six additional pathways are also recovered from the same experimental block, without increasing the probe duty cycle. These experiments give intra- and inter residue connectivities for the backbone \(^{13}{\text {C}}_\alpha\), \(^{15}{\text {N}}\), \(^{1}{\text {H}}_{\text {N}}\) and \(^{1}{\text {H}}_\alpha\) resonances and should alone be sufficient to assign these nuclei in proteins at MAS frequencies > 60 kHz. The validity of this approach is tested with experiments on a standard tripeptide N-formyl methionyl-leucine-phenylalanine (f-MLF) at a MAS frequency of 62.5 kHz, which is also used as a test-case for determining the sensitivity of each of the experiments. We expect this approach to have an immediate impact on the way assignments are obtained at MAS frequencies \(> 60\text { kHz}\).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Magic-angle-spinning solid-state NMR employing spinning frequencies \(>{\text {60 kHz}}\) is rapidly emerging as a viable technique for understanding the structural biology of several classes of proteins, including microcrystalline proteins (Knight et al. 2011; Agarwal et al. 2014; Andreas et al. 2016), membrane proteins (Linser et al. 2011b; Ward et al. 2011; Barbet-Massin et al. 2014; Dannatt et al. 2015; Lakomek et al. 2017), aggregated proteins and supramolecular assemblies (Marchetti et al. 2012; Struppe et al. 2017). Methodological developments in the study of dynamics of such proteins (Smith et al. 2017; Asami and Reif 2017; Smith et al. 2018; Rovó et al. 2019; Jain et al. 2019), as well as experiments that provide long and short-range distance restraints (Jain et al. 2017; Duong et al. 2018), promise to provide a complete picture of their structure and function. These developments, including assignment techniques have been reviewed elsewhere (Andreas et al. 2015a; Higman 2018). Despite the improvements in the resolution of \(^{1}{\text {H}}\) resonances in fully protonated proteins at MAS frequencies \(>{\text { 60 kHz}}\), obtaining site-specific assignments still remains the main bottleneck in these studies. The main reason for this is that the high resolution in the \(^{13}{\text {C}}\) and \(^{15}{\text {N}}\) dimensions alone is not sufficient to compensate for the comparatively low resolution in the \(^{1}{\text {H}}\) dimension in fully protonated proteins, improvements due to the ever-increasing MAS frequencies notwithstanding. With this background, the ideal 3D or 4D experiment that helps remove degeneracies in assignments is the residue-linking experiment \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\) (Andreas et al. 2015b; Xiang et al. 2015). This experiment encodes two dimensions with the chemical shift of a nucleus that has the highest resolution (\(^{15}{\text {N}}\)). In the best of conditions, this spectrum may alone be sufficient to generate complete assignments in small proteins. The \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\) is the \(^{13}{\text {C}}\)-edited analog of the above experiment. Work from the Pintacuda group suggests that H\(_\alpha\) resonances have lower line-widths than H\(_{\text {N}}\) in proteins (Stanek et al. 2016) and the inherent dependence of the \({\text {C}}_\alpha\) chemical shifts on amino-acid type makes this experiment a good candidate for assigning proteins as well. To the best of our knowledge, this experiment has not yet been reported for proteins, perhaps due to the higher resolution in the \(^{15}{\text {N}}\) dimension, which this strategy relies on, and the inherent insensitivity, which makes the acquisition of both these experiments unfeasible.
Both these strategies follow from the HNN experiment in solution NMR (Panchal et al. 2001), where the low dispersion in the \(^{1}{\text {H}}\) dimension for unstructured proteins is compensated by using two \(^{15}{\text {N}}\) dimensions with high resolution for assignments. When combined with the standard triple-resonance experiments such as \({\text {C}}_\alpha {\text {NH}}_{\text {N}}\) and \({\text {NC}}_\alpha {\text {H}}_\alpha\) (Linser et al. 2011b), these are expected to give complete assignments in even larger proteins. However, recording multiple 3D experiments in solids for the purpose of assignments is a time intensive task, requiring weeks of experimental time. One of the most promising approaches developed in the past few years has been the sequential detection of multiple experiments, which has been shown to allow 2–4 experiments to be detected in a single experimental block (Linser et al. 2011a; Gopinath and Veglia 2012b). This strategy makes use of either additional sources of polarization that are detected independently (Herbst et al. 2008; Gopinath and Veglia 2012a, b; Gopinath et al. 2019), or recovers polarization from coherence transfer pathways that are otherwise phase-cycled out in a traditional experiment (Banigan and Traaseth 2012; Mote et al. 2013; Gopinath et al. 2013; Gopinath and Veglia 2016). These strategies have also been adapted in oriented NMR studies of membrane proteins (Gopinath et al. 2015). They have been extended to \(^{1}{\text {H}}\) detected experiments (Sharma et al. 2016; Das and Opella 2016), where the presence of a third nucleus for correlation increases dramatically the number of ways in which multiple experiments can be combined. Fast MAS frequencies and \(^{1}{\text {H}}\) detection also allow for a comparatively higher efficiency of transfers (Penzel et al. 2015) and the aforementioned residue-linking experiments become feasible (Andreas et al. 2015a; Xiang et al. 2015) under these conditions. However, with the presence of multiple coherence transfers, the number of coherence transfer pathways that are discarded in the course of traditional phase cycling also increases.
Using the strategies gleaned from those used in experiments that involve sequential detection, we present here an approach that does the following two things: (i) simultaneously record the forward and backward residue-linking experiments \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\) and \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\), and (ii) recovers spectra from six other pathways that give intra- and inter-residue connectivities, which are otherwise discarded. A unique aspect of this experiment is that these gains are achieved with a single direct acquisition and no increase in the probe duty cycle, which avoid some of the problems associated with multiple sequential detection strategies. This results in simultaneous recording of eight complementary 2D experiments or six 3D intra- and inter-residue-linking experiments that are expected to give complete assignments in small to medium sized proteins, at MAS frequencies \(>{\text { 60 kHz}}\). A proof-of-principle demonstration is shown on a standard tripeptide MLF at a MAS frequency of 62.5 kHz. The availability of multiple spectra for assignments also improves the ability of automated assignment protocols such as ssFLYA (Schmidt et al. 2013), to improve the accuracy of assignments. In this context, the \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\) experiment is expected to be very useful in assigning proteins and further improve the resolution of degeneracies in automated assignment procedures. Given that CP based transfer efficiencies are expected to remain constant for a wide variety of samples in the solid state, we anticipate that this strategy will be a very efficient way to address the assignment problem for proteins at MAS frequencies \(>{\text {60 kHz}}\).
Results and discussion
The pulse sequence
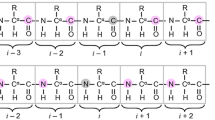
Figure 1 shows the pulse sequence which combines \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\) and \({\text {C}}_\alpha\)(CoN)\({\text {C}}_\alpha\)H\(_\alpha\) pulse sequences as in previously described multiple-acquisition experiments (Gopinath and Veglia 2012b, 2013; Sharma et al. 2016; Kupče et al. 2019). The \(^{1}{\text {H}}\) magnetization is initially transferred to \(^{13}{\text {C}}\) and \(^{15}{\text {N}}\) using simultaneous cross-polarization (Herbst et al. 2008; Linser et al. 2011a; Gopinath and Veglia 2012b) and the \(^{13}{\text {C}}_\alpha\) and the amide \(^{15}{\text {N}}\) nuclei are encoded in the first indirect dimension. A series of transfer steps, starting with \({\text {C}}_\alpha \rightarrow {\text {Co}}\) (DREAM), followed by Co \(\leftrightarrow\)N and Co \(\rightarrow {\text {C}}_\alpha\) (DREAM), and finally N\(_\alpha\)\(\leftrightarrow {\text {C}}_\alpha\) leave the polarization originating on \(^{13}{\text {C}}_\alpha\) nucleus on the \(^{13}{\text {C}}_\alpha\) nucleus of the next residue, while the polarization originating on the amide \(^{15}{\text {N}}\) nucleus now resides on the amide \(^{15}{\text {N}}\) nucleus of the preceding residue. The chemical shifts of these nuclei are now encoded in the second indirect dimension during a 3D experiment. Finally, the polarization is transferred to \(^{1}{\text {H}}\) for detection after a water-suppression element. The \(^{15}{\text {N}}\) polarization is kept along the longitudinal axis during the course of the DREAM transfers. This design ensures that there is no increase in the probe duty cycle. In cases of highly resolved spectra of small proteins, the \({\text {N(CoC}_\alpha)}{\text {NH}}_{\text {N}}\) experiment is alone expected to give nearly complete assignments (Andreas et al. 2016; Stanek et al. 2016). Given the inherent dependence of the \({\text {C}}_\alpha\) chemical shift on the amino-acid type and the high resolution for the H\(_\alpha\) resonances, we expect the \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\) experiment to perform similarly well for this purpose. This sequence can be modified with selective \(\pi\) pulses on the carbonyl nuclei during the indirect evolution times for achieving scalar decoupling of both \(^{15}{\text {N}}\) and \(^{13}{\text {C}}_\alpha\) nuclei. However, if the \(^{15}{\text {N}}\)–\(^{13}{\text {C}}_\alpha\) scalar coupling is to be refocused, the two nuclei will need to have independent evolution periods. This can be easily achieved by storing \(^{15}{\text {N}}\) polarization along the longitudinal direction during \(^{13}{\text {C}}\) evolution, and vice-versa (Gopinath and Veglia 2013; Sharma et al. 2016). Note that this increases the duty cycle compared to the standard experiment. It is also possible to use INEPT instead of the DREAM to achieve \({\text {C}}_\alpha\)\(\leftrightarrow\) Co transfers. The relative merits of using INEPT and DREAM depend on the MAS frequency, and these issues have been discussed elsewhere (Barbet-Massin et al. 2013; Penzel et al. 2015; Fraga et al. 2017). \(^{15}{\text {N}}\) and \(^{13}{\text {C}}\) nuclei in proteins exhibit different \({\text {T}}_2\) relaxation and chemical-shift dispersions, and are often recorded with different spectral widths and indirect evolution times. In the design where \(^{15}{\text {N}}\) and \(^{13}{\text {C}}\) are co-evolved, an arbitrary setting of spectral width and evolution times can be achieved by first storing both polarizations along the longitudinal axis, and then recalling them with \(\pi /2\) pulses in such a way that the evolution times for a particular indirect point end at the same time, as has been shown before (Gopinath and Veglia 2013; Sharma et al. 2016).

a Pulse sequence for the acquisition of multiple \(^{1}{\text {H}}\) detected experiments in a single experiment. The pathway for the acquisition of the \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\) and \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\) experiments are shown by yellow and pink arrows, respectively. b Colour-coded pathways for all the experiments encoded by this pulse sequence. The start and the end of each pathway on either \(^{1}{\text {H}}_\alpha\) or \(^{1}{\text {H}}_{\text{N}}\) are indicated by arrows. The numbers that represent each of the pathways are 1: \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\), 2: \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\), 3: N(Co)\({\text {C}}_\alpha\) H\(_\alpha\), 4: \({\text {C}}_\alpha {\text {(Co)NH}}_{\text {N}}\), 5: \({\text {NC}}_\alpha {\text {H}}_\alpha\), 6: \({\text {C}}_\alpha {\text {NH}}_{\text {N}}\), 7: NH\(_{\text {N}}\), and 8: CxHx. Narrow bars indicate 90° pulses and the pulse blocks are labelled as follows: Sim-CP: simultaneous cross-polarization (Herbst et al. 2008; Gopinath and Veglia 2012b), DREAM: dipolar recoupling using adiabatic modulation (Verel et al. 2001), SP-CP: specific cross-polarization using the double-quantum-recoupling conditions, and \({\text {rCW}^{ApA}}\): refocused continuous-wave heteronuclear decoupling, using conditions optimized for 62.5 kHz MAS and low RF (Equbal et al. 2017). The 16-step phase cycle is: \(\phi _1=\text {y}\), − y; \(\phi _2=\text {x}\), \(\phi _3=\text {y}\), \(\phi _4=-\text {y}\), \(\phi _5=\text {x}\), x, − x, − x; \(\phi _6=\phi _7=\text {x}\), \(\phi _8=\text {(x)}\times 4\), (− x) × 4; \(\phi _9=\text {(x)}\times 8\), (− x) × 8; \(\phi _{10}=\text {(x)}\times 8\), (− x) × 8. This can be increased to a 32-step cycle by using \(\phi _{10}=\text {(x)}\times 16\), (− x) × 16 or reduced to an 8-step cycle using \(\phi _9=\phi _{10}=\text {x}\) (see main text for justification). The phase cycle on the initial \(^{1}{\text {H}}\) pulse may also be not necessary in most samples, reducing the minimum phase cycle required for this experiment to 4. One can impose additional phase cycles on \(\phi _2\) and the \(\pi /2\) pulses flanking the water-suppression block. This is generally not required, but is recommended if the number of scans used are 64, or higher. The phases \(\phi _5\), \(\phi _8\), \(\phi _9\) and \(\phi _{10}\) are used in the multiplex phase cycle (see main text for more details), and consequently, the receiver phase \(\phi _{rec}\) (x, − x) follows only the \(^{1}{\text {H}}\)\(90^\circ\) pulse phase in absence of additional phase cycle on \(\phi _2\), \(\phi _3\), \(\phi _4\), \(\phi _6\) and \(\phi _7\). Note that the cross-polarization from \(^{15}{\text {N}}\) to \(^{1}{\text {H}}\) and \(^{13}{\text {C}}\) to \(^{1}{\text {H}}\) uses different contact times, and the pulse blocks are arranged so that both of them end at the same time (Sharma et al. 2016; Das and Opella 2016). Quadrature detection for the indirect dimensions is achieved using the STATES-TPPI method for the pulses marked with an asterix
Recovering orphan pathways using multiplexing
At this point, a simple 8 or 16-step phase cycle can be used to select out the desired pathways, the minimum being a 4-step phase cycle (vide infra). A 16-step phase cycle that achieves this is given in Fig. 1. The most prominent alternate pathways that are cancelled out during the course of this phase cycling arise due to the inefficiency of \(^{15}{\text {N}}\)\(\leftrightarrow\)\(^{13}{\text {C}}\) transfers. There are pathways that result from polarization not transferred at the N \(\leftrightarrow\) Co step (\({\text {C}}_\alpha\)NH and \({\text {NC}}_\alpha {\text {H}}_\alpha\)), the N \(\leftrightarrow {\text {C}}_\alpha\) step (\({\text {C}}_\alpha\)(Co)NH, N(Co)\({\text {C}}_\alpha\)H\(_\alpha\)), or both the steps (\({{{\text {C}}_\alpha}{{\text {C}}_\alpha}}\)H\(_\alpha\) and \({\text {NNH}}_{\text {N}}\)). As their names suggest, these pathways can complement the residue-linking experiments by providing additional intra- and inter-residue connectivities. They can be selected from the pulse sequence in Fig. 1 with an identical phase cycle as long as the receiver phase is adjusted for the pathway of interest. Here, we recover all of these pathways, and make use of multiplexing (Ivchenko et al. 2003) to do so. Each individual free-induction-decay (FID) resulting from the phase cycle is stored separately and then multiplied with an appropriate phase factor before adding all of the FIDs to give the signal from the desired pathway. Thus, the same dataset processed with eight different combinations of phase factors will result in 8 independent spectra. One needs to only ensure that selecting out one particular pathway completely cancels out all other pathways. This can be done using the phase cycle and the deconvolution matrix shown in Fig. 2. The resulting experimental spectra on the tripeptide f-MLF are shown in Fig. 3. Here, the indicated 2D experiments were acquired using the pulse sequence in Fig. 1 where only the first indirect dimension (\({\text {t}}_1\)) is incremented. We note that this strategy has been previously used to recover two (Gopinath et al. 2013) and four (Sharma et al. 2016) experiments from a single direct acquisition. This article extends this to the recovery of eight experiments. In general, the number of pathways in any experiment is expected to be four times the number of \(^{15}{\text {N}}\)\(\leftrightarrow\)\(^{13}{\text {C}}\) SPECIFIC-CP transfers (two of them being residual pathways, which arise due to the inefficiency of the CP block). The use of two CP blocks in the residue-linking experiments, thus, lends itself naturally to multiplexing and the recovery of eight experiments. When MAS frequencies > 60 kHz and \(^{1}{\text {H}}\) detection are used, this recovery is achieved using a single direct acquisition and no increase in probe duty cycle, which is not possible at slower MAS frequencies and \(^{13}{\text {C}}\) detection. It may be possible to record this experiment as a series of direct acquisitions, and even possible to recover residual polarization after the final N \(\rightarrow\) H\(_{{\text {N}}}\) and C \(\rightarrow\) H\(_\alpha\) transfers. However, each additional \(^{1}{\text {H}}\) detection element is expected to require an additional water-suppression block. Even a short water-suppression block of 40–50 ms adds to the overall experimental time (~ 2–3% increase in time), and causes a net sensitivity loss of 16–20%, negating in part the gains on some of the polarization pathways, especially those with low sensitivity. Thus, the use of a single detection and a single water-suppression element is critical to the recovery of these pathways. We note that experiments recovered here form the basis of assignment strategies at MAS frequencies > 60 kHz, and would be required for complete assignments of backbone atoms using \(^{1}{\text {H}}\) detected experiments under these conditions, even if the strategy outlined here is not used.

Multiplex phase-cycling to separate out 8 independent pathways. 16 1D experiments arranged as a matrix (a), when multiplied by the deconvolution matrix (b), give a set of 8 1D datasets (c), which correspond to eight independent pathways shown in (d). White bars in the second column indicate a phase of + x, while black bars indicate a phase of − x. Each 1D (Column a) is a combination of two scans, with \(\phi _1=\text {y}\), − y and \(\phi _{rec}=\text {x}\), − x, where the phases refer to those described in Fig. 1. The phases that are a part of the multiplex phase cycle are \(\phi _5\), \(\phi _8\), \(\phi _9\) and \(\phi _{10}\), with a nested phase cycle x, − x on each of them. Each decounvoluted 1D spectrum c is, thus, a summation of 32 scans. Red and blue vertical lines in Column c are used to distinguish the positions of the three H\(_{\text {N}}\) and H\(_\alpha\) resonances, respectively

(a–h) Eight 2D experiments recorded on f-MLF using the pulse sequence shown in Fig. 1. The indirect dimension evolved in this experiment is denoted by \({\text {t}}_1\) in Fig. 1. 38 complex points were acquired in the indirect dimension, corresponding to an evolution time of 7.6 ms for both \(^{13}{\text {C}}\) and \(^{15}{\text {N}}\). Side-chain peaks in the spectra from pathway 8 (CxHx) are indicated by an asterix in h
Sensitivity of the eight pathways
It is instructive to compare the sensitivity of each of these experiments in order to assess the overall time savings. The sensitivity of each of the experiments is linked to the efficiency of each of the transfer blocks. At 62.5 kHz on the f-MLF sample, we have found that the efficiencies of each of the transfer steps is as follows: NCo: 0.50, NC\(_\alpha\): 0.38, \({\text {C}}_\alpha\) Co: 0.41, CoC\(_\alpha\): 0.43. The \(^{15}{\text {N}}\)\(\leftrightarrow\)\(^{13}{\text {C}}\) transfer efficiencies were determined by comparing the sensitivity of the \(^{15}{\text {N}}\) cross-polarization experiment versus the sensitivity of \(^{15}{\text {N}}\)\(\rightarrow\)\(^{13}{\text {C}}\)\(\rightarrow\)\(^{15}{\text {N}}\) experiment (the net efficiency of a single transfer is given by the square root of the ratio of the SNR in these experiments), as described by Penzel et al. (2015). The efficiency of DREAM transfers was determined by comparing the integral of the \(^{13}{\text {C}}_\alpha\) resonances after the DREAM transfer to that of the directly-bonded \(^{13}{\text {Co}}\) resonance before the transfer, and vice-versa. These numbers are close to those reported for ubiquitin at a MAS frequency of \(\sim 100\text { kHz}\) (Penzel et al. 2015), and are likely to improve mainly due to the higher efficiency of \({\text {C}}_\alpha\)-Co DREAM transfers at MAS frequencies > 100 kHz. The fraction of the overall magnetization available for each individual experiment with respect to that available in a pulse sequence optimized for each of these experiments is given in Table 1—Column A. These numbers are given with respect to a pulse sequence that is optimized for a single experiment. For example, the \({\text {C}}_\alpha {\text {NH}}_{\text {N}}\) is compared with an experiment that does not use out-and-back DREAM transfers, as is mandated by the design of the pulse sequence in Fig. 1. Considering these numbers, we conclude that if each of these experiments are to be acquired individually, with the sensitivity that is obtained from the pulse sequence in Fig. 1, it would require an experimental time that is \(\sim 2.3\) times longer (This number is given by the sum of the squares of the numbers in Table 1—Column A). Note that since a minimum of 4-step phase cycle is required for the successful application of the above strategy (vide infra), practical time savings in case of high sensitivity are higher, as the overall experimental time is limited by the phase cycle of the remaining experiments. The use of multiple transfer steps in the residue-linking experiments makes it quite insensitive (Table 1—Column B), and a higher number of scans will be required to record this experiment with adequate signal-to-noise. This means that the other 6 experiments, despite the low values of the initial polarization available, end up with enough sensitivity for a reliable detection of resonances (Table 1—Column C). However, when the DREAM and/or SPECIFIC-CP transfer efficiencies are significantly lower than those reported here, a prohibitively long time may be required for the residue-linking experiments. It will then be possible to record experiments such as \({\text {NC}}_\alpha {\text {H}}_\alpha\) and \({\text {C}}_\alpha {\text {NH}}_{\text {N}}\) which require a lower number of transfer steps, with a lower number of scans than those used to calculate the relative time savings here. In such cases, it will be important to consider the minimum number of scans with which each experiment can be done, rather than the net sensitivity, and the overall time savings are expected to be lower than the factor of 2.3 that we report.
The minimum phase cycle
The minimum phase cycle to cleanly separate all eight experiments has to be at least eight steps long, thus mandating a minimum of 8 scans for this experiment. The simplest way to achieve this is to nest 2-step phase cycles (x, − x) on the two \(^{15}{\text {N}}\)\(\leftrightarrow\)\(^{13}{\text {C}}\) transfers and the final transfer to \(^{1}{\text {H}}\) from either \(^{13}{\text {C}}\) or \(^{15}{\text {N}}\). Such a phase cycle for each of the eight pathways is shown in Figs. 1 and 2. The addition of an independent 2-step phase cycle (y, − y) for the first \(^{1}{\text {H}}\) 90° pulse to suppress undesired pathways starting with initial \(^{13}{\text {C}}\) or \(^{15}{\text {N}}\) polarizations doubles the minimum number of scans per increment to 16. On our 1.3 mm HCN probe, the large background signal from \(^{1}{\text {H}}\) in one of the pathways (\({\text {NNH}}_{{\text {N}}}\)) needed to be suppressed by an additional 2-step phase cycle, bringing the net number of scans required per 2D experiment to 32. However, the \(^{1}{\text {H}}_\alpha\) and \(^{1}{\text {H}}_{{\text {N}}}\) resonances occupy different parts of the spectral window, and hence, are already separated in the direct dimension. If their separation is deemed unnecessary, the total number of scans can be reduced to 16. The phase cycle on the initial \(^{1}{\text {H}}\) pulse can also be removed as the contribution from longitudinal \(^{15}{\text {N}}\) and \(^{13}{\text {C}}\) magnetization is small or is dephased prior to the pulse. Further, in 3D (and higher-dimensional experiments), the \({\text {NNH}}_{\text {N}}\) pathway does not contain more information than the corresponding 2D experiment, and can be ignored completely. This brings the absolute minimum number of scans required to implement this strategy down to 4. It may be possible to record this experiment with only 4 scans on small proteins with exceptional sensitivity, but most samples are expected to require a higher number of scans, and it may be prudent to include additional phase-cycling blocks, including the ones mentioned above, in the final phase cycle.
Extensions to higher dimensions
The extension of this experiment to three or higher dimensions can be done in a straightforward manner using chemical-shift evolution periods at appropriate positions in the pulse sequence shown in Fig. 1. 3D cubes of the eight 3D experiments obtained on the f-MLF sample are shown in Fig. 4. It is to be noted that the residual CxCxHx and \({\text {NNH}}_{\text {N}}\) experiments have a repeated dimension, and thus have no more information than the corresponding 2D dataset. However, in situations where slow chemical exchange occurs between one or more conformations with distinct \(^{15}{\text {N}}\) chemical shifts, the \({\text {NNH}}_{\text {N}}\) experiment may give off-diagonal peaks in the \(^{15}{\text {N}}\)–\(^{15}{\text {N}}\) plane (Cho et al. 2014). This can be used for diagnostic purposes as well as to prevent misassignments. We note that a single 5D-NCoC\(_\alpha\)NH\(_{\text {N}}\) experiment was recently shown to give nearly complete assignments in medium-sized proteins (Orton et al. 2019). This experiment uses a coherence transfer pathway that is similar to the \({\text {N(CoC}}_\alpha){\text {NH}}_{\text {N}}\) pathway shown in Fig. 1. The addition of information from 5 complementary pathways using the strategy outlined above is expected to speed up the assignment process for proteins even further. Experiments with >3 dimensions are expected to require the use of non-uniform sampling (NUS) (Mobli and Hoch 2014). The strategy outline above is compatible with NUS; the preprocessing of time-domain data as described in Fig. 2 will result in multiple datasets that can be then be processed using any of the available NUS reconstruction algorithms.

(a–h) Eight 3D experiments recorded using the pulse sequence shown in Fig. 1 on f-MLF. These experiments were recorded with 32 scans and 16 complex points in each indirect dimension, corresponding to an evolution time of 3.2 ms in each indirect dimension
Materials and methods
NMR experiments were done on a 16.5 T Bruker magnet (\(\sim 700\text { MHz}\)\(^{1}{\text {H}}\) Larmor frequency) with a 1.3 mm HCN triple resonance probe (Bruker Biospin) controlled using an Avance III console and Topspin 3.5. All experiments were done at a MAS frequency of 62.5 kHz. Cross-polarization from \(^{1}{\text {H}}\) to \(^{13}{\text {C}}\) and \(^{15}{\text {N}}\) was done using the double-quantum recoupling condition (\(\nu _H\sim 50\text { kHz}\), and \(\nu _C\) and \(\nu _N\sim 12.5\text { kHz}\)). These conditions avoid the known PAR/PAIN recoupling conditions (Agarwal et al. 2013). Contact times for CP were optimized independently and the best contact time for the transfer to \(^{13}{\text {C}}\) was chosen. This gave \(\sim 80\%\) transfer to \(^{15}{\text {N}}\) nuclei, similar to the transfer seen at slow-moderate MAS frequencies (Gopinath and Veglia 2012b), while the transfer to \({\text {C}}_\alpha\) was reduced to \(\sim 0.93\) times its original value. The DREAM sequence with adiabatic modulation of the radio-frequency (RF) amplitude (Verel et al. 2001) was used for transfer of polarization from \(^{13}{\text {Co}}\) to \(^{13}{\text {C}}_\alpha\) and vice-versa (\(\nu _C\sim 31\text { kHz}\); RF offset placed between \(^{13}{\text {Co}}\) and \(^{13}{\text {C}}_\alpha\), both optimized independently). Transfers from \(^{15}{\text {N}}\) to \(^{13}{\text {Co}}\) and \(^{13}{\text {C}}_\alpha\), and vice-versa were optimized at the double-quantum transfer conditions using tangential modulation of the RF amplitude on \(^{13}{\text {C}}\) channel (\(\nu _N\sim 50\text { kHz}\) and \(\nu _C\sim 12.5\text { kHz}\)). Water suppression was achieved using the MISSISSIPI sequence Zhou and Rienstra (2008); only 10 ms long block was used for the dry f-MLF sample. The contact times for the reverse cross polarization to \(^{1}{\text {H}}\) were \(150\,\upmu {\text {s}}\) and \(50\,\upmu {\text {s}}\) for \(^{15}{\text {N}}\) and \(^{13}{\text {C}}\), respectively. Low-power \({\text {rCW}^{ApA}}\) decoupling (Equbal et al. 2017) was used in the indirect dimensions, and 15 ms \(^{1}{\text {H}}\) detection was carried out under WALTZ-16 heteronuclear decoupling (Shaka et al. 1983) on both \(^{13}{\text {C}}\) and \(^{15}{\text {N}}\) channels. Spectra were processed using NMRGlue (v0.8-dev) (Helmus and Jaroniec 2013) and Topspin 3.5, and figures were generated using Matploltib and Inkscape. 3D box plots were made using nmrcube routine in NMRPipe (Delaglio et al. 1995). The pulse sequence and instructions for processing the spectra are included as supplemantary information to this article. These will be additionally made available at www.github.com/kaustubhmote/pulseseq, and can also be requested from the authors by email.
Conclusions
We have demonstrated here the utility of multiplex phase cycling to acquire as many as eight 2D and six 3D experiments within a single experimental block, using a single direct acquisition. This method uses strategies that were developed for experiments that utilize multiple sequential acquisition of coherence pathways, and takes advantage of the higher sensitivity of \(^{1}{\text {H}}\) detection, which allows such a combination to be recorded. They give complementary intra- and inter-residue connectivities that will allow for a direct assignment of the backbone residues. The use of a \(^{13}{\text {C}}\)-edited residue-liking experiment, namely \({\text {C}}_\alpha {\text {(CoN)C}_\alpha} {\text {H}}_\alpha\), although anticipated due the success of \(^{15}{\text {N}}\)-edited residue-linking experiments \({\text {(N(C}}_\alpha {\text {Co)NH}}_{\text {N}})\) has not yet been demonstrated. The relative insensitivity of these experiments means that a choice needed to be made with respect to which experiment would be best suited. Instead, our strategy combines these two experiments and makes them available for assignments at the same time. The six additional 2D or four 3D datasets contain complementary intra- and inter-residue connectivities, which will help in resolving degeneracies for larger proteins. Notably, this is achieved without increasing the duty cycle of the standard pulse sequence and thus is applicable irrespective of the nature of the protein sample. The use of a single detection and a single water-suppression element also means that the gains from the recovery of some of the more insensitive pathways are retained, which would have otherwise been lost due to a perceptively small, but significant increase in experimental time. We anticipate that this 3D experiment, as well as higher dimensional experiments (which evolve the chemical shift of the \(^{13}{\text {Co}}\) resonances) will be sufficient to assign backbone resonances in proteins at MAS frequencies > 100 kHz.
References
Agarwal V, Sardo M, Scholz I, Böckmann A, Ernst M, Meier BH (2013) PAIN with and without PAR: variants for third-spin assisted heteronuclear polarization transfer. J Biomol NMR. https://doi.org/10.1007/s10858-013-9756-4
Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, Past J, Samoson A, Ernst M, Böckmann A, Meier BH (2014) De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed 53(45):12253–12256. https://doi.org/10.1002/anie.201405730
Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G (2015a) High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson 253:36–49. https://doi.org/10.1016/j.jmr.2015.01.003
Andreas LB, Stanek J, Marchand TL, Bertarello A, Paepe DCD, Lalli D, Krejčíková M, Doyen C, Öster C, Knott B, Wegner S, Engelke F, Felli IC, Pierattelli R, Dixon NE, Emsley L, Herrmann T, Pintacuda G (2015b) Protein residue linking in a single spectrum for magic-angle spinning NMR assignment. J Biomol NMR 62(3):253–261. https://doi.org/10.1007/s10858-015-9956-1
Andreas LB, Jaudzems K, Stanek J, Lalli D, Bertarello A, Le Marchand T, Cala-De Paepe D, Kotelovica S, Akopjana I, Knott B, Wegner S, Engelke F, Lesage A, Emsley L, Tars K, Herrmann T, Pintacuda G (2016) Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1602248113
Asami S, Reif B (2017) Comparative study of REDOR and CPPI derived order parameters by \(^1\)H-detected MAS NMR and MD simulations. J Phys Chem B 121(37):8719–8730. https://doi.org/10.1021/acs.jpcb.7b06812
Banigan JR, Traaseth NJ (2012) Utilizing afterglow magnetization from cross-polarization magic-angle-spinning solid-state NMR spectroscopy to obtain simultaneous heteronuclear multidimensional spectra. J Phys Chem B 116(24):7138–7144. https://doi.org/10.1021/jp303269m
Barbet-Massin E, Pell AJ, Jaudzems K, Franks WT, Retel JS, Kotelovica S, Akopjana I, Tars K, Emsley L, Oschkinat H et al (2013) Out-and-back 13C–13C scalar transfers in protein resonance assignment by proton-detected solid-state nmr under ultra-fast mas. J Biomol NMR 56(4):379–386
Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, Nieuwkoop AJ, Hiller M, Higman V, Guerry P, Bertarello A, Knight MJ, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou JJ, Griffin RG, Oschkinat H, Lesage A, Emsley L, Herrmann T, Pintacuda G (2014) Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc 136(35):12489–12497. https://doi.org/10.1021/ja507382j
Cho MK, Gayen A, Banigan JR, Leninger M, Traaseth NJ (2014) Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J Am Chem Soc 136(22):8072–8080. https://doi.org/10.1021/ja503145x
Dannatt HRW, Taylor GF, Varga K, Higman VA, Pfeil MP, Asilmovska L, Judge PJ, Watts A (2015) 13C- and 1H-detection under fast MAS for the study of poorly available proteins: application to sub-milligram quantities of a 7 trans-membrane protein. J Biomol NMR 62(1):17–23. https://doi.org/10.1007/s10858-015-9911-1
Das BB, Opella SJ (2016) Simultaneous cross polarization to 13C and 15N with 1H detection at 60kHz MAS solid-state NMR. J Magn Reson 262:20–26. https://doi.org/10.1016/j.jmr.2015.12.004
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6(3):277–293. https://doi.org/10.1007/bf00197809
Duong NT, Raran-Kurussi S, Nishiyama Y, Agarwal V (2018) Quantitative 1H–1H distances in protonated solids by frequency-selective recoupling at fast magic angle spinning NMR. J Phys Chem Lett 9(20):5948–5954. https://doi.org/10.1021/acs.jpclett.8b02189
Equbal A, Madhu PK, Meier BH, Nielsen NC, Ernst M, Agarwal V (2017) Parameter independent low-power heteronuclear decoupling for fast magic-angle spinning solid-state NMR. J Chem Phys 146(8):084202. https://doi.org/10.1063/1.4976997
Fraga H, Arnaud CA, Gauto DF, Audin M, Kurauskas V, Macek P, Krichel C, Guan JY, Boisbouvier J, Sprangers R et al (2017) Solid-state NMR H-N-(C)-H and H-N-C-C 3D/4D correlation experiments for resonance assignment of large proteins. ChemPhysChem 18(19):2697–2703
Gopinath T, Veglia G (2012a) 3D DUMAS: simultaneous acquisition of three-dimensional magic angle spinning solid-state NMR experiments of proteins. J Magn Reson 220:79–84. https://doi.org/10.1016/j.jmr.2012.04.006
Gopinath T, Veglia G (2012b) Dual acquisition magic-angle spinning solid-state NMR-spectroscopy: simultaneous acquisition of multidimensional spectra of biomacromolecules. Angew Chem Int Ed 51(11):2731–2735. https://doi.org/10.1002/anie.201108132
Gopinath T, Veglia G (2013) Orphan spin operators enable the acquisition of multiple 2D and 3D magic angle spinning solid-state NMR spectra. J Chem Phys 138(18):184201. https://doi.org/10.1063/1.4803126
Gopinath T, Veglia G (2016) Multiple acquisitions via sequential transfer of orphan spin polarization (MAeSTOSO): how far can we push residual spin polarization in solid-state NMR? J Magn Reson 267:1–8. https://doi.org/10.1016/j.jmr.2016.03.001
Gopinath T, Mote KR, Veglia G (2013) Sensitivity and resolution enhancement of oriented solid-state NMR: application to membrane proteins. Prog Nucl Magn Reson Spectrosc 75:50–68. https://doi.org/10.1016/j.pnmrs.2013.07.004
Gopinath T, Mote KR, Veglia G (2015) Simultaneous acquisition of 2D and 3D solid-state NMR experiments for sequential assignment of oriented membrane protein samples. J Biomol NMR 62(1):53–61. https://doi.org/10.1007/s10858-015-9916-9
Gopinath T, Wang S, Lee J, Aihara H, Veglia G (2019) Hybridization of TEDOR and NCX MAS solid-state NMR experiments for simultaneous acquisition of heteronuclear correlation spectra and distance measurements. J Biomol NMR 73(3–4):141–153. https://doi.org/10.1007/s10858-019-00237-5
Helmus JJ, Jaroniec CP (2013) Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J Biomol NMR 55(4):355–367. https://doi.org/10.1007/s10858-013-9718-x
Herbst C, Riedel K, Ihle Y, Leppert J, Ohlenschläger O, Görlach M, Ramachandran R (2008) MAS solid state NMR of RNAs with multiple receivers. J Biomol NMR 41(3):121–125. https://doi.org/10.1007/s10858-008-9247-1
Higman VA (2018) Solid-state MAS NMR resonance assignment methods for proteins. Prog Nucl Magn Reson Spectrosc 106:37–65
Ivchenko N, Hughes CE, Levitt MH (2003) Multiplex phase cycling. J Magn Reson 160(1):52–58. https://doi.org/10.1016/S1090-7807(02)00108-8
Jain MG, Lalli D, Stanek J, Gowda C, Prakash S, Schwarzer TS, Schubeis T, Castiglione K, Andreas LB, Madhu PK, Pintacuda G, Agarwal V (2017) Selective \(^1{\rm H}-^1{\rm H}\) distance restraints in fully protonated proteins by very fast magic-angle spinning solid-state NMR. J Phys Chem Lett 8(11):2399–2405. https://doi.org/10.1021/acs.jpclett.7b00983
Jain MG, Mote KR, Hellwagner J, Rajalakshmi G, Ernst M, Madhu PK, Agarwal V (2019) Measuring strong one-bond dipolar couplings using REDOR in magic-angle spinning solid-state NMR. J Chem Phys 150(13):134201. https://doi.org/10.1063/1.5088100
Knight MJ, Webber AL, Pell AJ, Guerry P, Barbet-Massin E, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Emsley L, Lesage A, Herrmann T, Pintacuda G (2011) Fast resonance assignment and fold determination of human superoxide dismutase by high-resolution proton-detected solid-state MAS NMR spectroscopy. Angew Chem Int Ed 50(49):11697–11701. https://doi.org/10.1002/anie.201106340
Kupče E, Mote KR, Madhu PK (2019) Experiments with direct detection of multiple FIDs. J Magn Reson 304:16–34. https://doi.org/10.1016/j.jmr.2019.04.018
Lakomek NA, Frey L, Bibow S, Böckmann A, Riek R, Meier BH (2017) Proton-detected NMR spectroscopy of nanodisc-embedded membrane proteins: MAS solid-state vs. solution-state methods. J Phys Chem B 121:7671–7680. https://doi.org/10.1021/acs.jpcb.7b06944
Linser R, Bardiaux B, Higman V, Fink U, Reif B (2011a) Structure calculation from unambiguous long-range amide and methyl 1H–1H distance restraints for a microcrystalline protein with MAS solid-state NMR spectroscopy. J Am Chem Soc 133(15):5905–5912. https://doi.org/10.1021/ja110222h
Linser R, Dasari M, Hiller M, Higman V, Fink U, Lopez Del Amo JMJM, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, Reif B (2011b) Proton-detected solid-state NMR spectroscopy of fibrillar and membrane proteins. Angew Chem Int Ed 50(19):4508–4512. https://doi.org/10.1002/anie.201008244
Marchetti A, Jehle S, Felletti M, Knight MJ, Wang Y, Xu ZQ, Park AY, Otting G, Lesage A, Emsley L, Dixon NE, Pintacuda G (2012) Backbone assignment of fully protonated solid proteins by 1H detection and ultrafast magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed 51(43):10756–10759. https://doi.org/10.1002/anie.201203124
Mobli M, Hoch JC (2014) Nonuniform sampling and non-Fourier signal processing methods in multidimensional NMR. Prog Nucl Magn Reson Spectrosc 83:21–41. https://doi.org/10.1016/j.pnmrs.2014.09.002
Mote KR, Gopinath T, Veglia G (2013) Determination of structural topology of a membrane protein in lipid bilayers using polarization optimized experiments (POE) for static and MAS solid state NMR spectroscopy. J Biomol NMR 57(2):91–102. https://doi.org/10.1007/s10858-013-9766-2
Orton HW, Stanek J, Schubeis T, Foucaudeau D, Ollier C, Draney AW, Le Marchand T, De Paepe DC, Felli IC, Pierattelli R et al (2019) Protein NMR resonance assignment without spectral analysis: 5D solid-state automated projection spectroscopy (SO-APSY). Angew Chem Int Ed. https://doi.org/10.1002/anie.201912211
Panchal SC, Bhavesh NS, Hosur RV (2001) Improved 3D triple resonance experiments, HNN and HN(C)N, for HN and 15N sequential correlations in (13C, 15N) labeled proteins: application to unfolded proteins. J Biomol NMR 20(2):135–147. https://doi.org/10.1023/A:1011239023422
Penzel S, Smith AA, Agarwal V, Hunkeler A, Org MLL, Samoson A, Böckmann A, Ernst M, Meier BH (2015) Protein resonance assignment at MAS frequencies approaching 100 kHz: a quantitative comparison of J-coupling and dipolar-coupling-based transfer methods. J Biomol NMR 63(2):165–186. https://doi.org/10.1007/s10858-015-9975-y
Rovó P, Smith CA, Gauto D, de Groot BL, Schanda P, Linser R (2019) Mechanistic insights into microsecond time-scale motion of solid proteins using complementary 15n and 1h relaxation dispersion techniques. J Am Chem Soc 141(2):858–869. https://doi.org/10.1021/jacs.8b09258
Schmidt E, Gath J, Habenstein B, Ravotti F, Székely K, Huber M, Buchner L, Böckmann A, Meier BH, Güntert P (2013) Automated solid-state NMR resonance assignment of protein microcrystals and amyloids. J Biomol NMR 56(3):243–254
Shaka AJ, Keeler J, Frenkiel T, Freeman R (1983) An improved sequence for broadband decoupling: WALTZ-16. J Magn Reson 52(2):335–338. https://doi.org/10.1016/0022-2364(83)90207-X
Sharma K, Madhu PK, Mote KR (2016) A suite of pulse sequences based on multiple sequential acquisitions at one and two radiofrequency channels for solid-state magic-angle spinning NMR studies of proteins. J Biomol NMR 65(3–4):127–141. https://doi.org/10.1007/s10858-016-0043-z
Smith AA, Ernst M, Meier BH (2017) Because the light is better here: correlation-time analysis by NMR spectroscopy. Angew Chem Int Ed 56(44):13590–13595. https://doi.org/10.1002/anie.201707316
Smith AA, Ernst M, Meier BH (2018) Optimized “detectors” for dynamics analysis in solid-state NMR. J Chem Phys 148(4):045104. https://doi.org/10.1063/1.5013316
Stanek J, Andreas LB, Jaudzems K, Cala D, Lalli D, Bertarello A, Schubeis T, Akopjana I, Kotelovica S, Tars K, Pica A, Leone S, Picone D, Xu ZQ, Dixon NE, Martinez D, Berbon M, El Mammeri N, Noubhani A, Saupe S, Habenstein B, Loquet A, Pintacuda G (2016) NMR spectroscopic assignment of backbone and side-chain protons in fully protonated proteins: microcrystals, sedimented assemblies, and amyloid fibrils. Angew Chem Int Ed 55(50):15504–15509. https://doi.org/10.1002/anie.201607084
Struppe J, Quinn CM, Lu M, Wang M, Hou G, Lu X, Kraus J, Andreas LB, Stanek J, Lalli D, Lesage A, Pintacuda G, Maas W, Gronenborn AM, Polenova T (2017) Expanding the horizons for structural analysis of fully protonated protein assemblies by NMR spectroscopy at MAS frequencies above 100 kHz. Solid State Nucl Magn Reson 87(June):117–125. https://doi.org/10.1016/j.ssnmr.2017.07.001
Verel R, Ernst M, Meier BH (2001) Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J Magn Reson 150(1):81–99. https://doi.org/10.1006/jmre.2001.2310
Ward ME, Shi L, Lake E, Krishnamurthy S, Hutchins H, Brown LS, Ladizhansky V (2011) Proton-detected solid-state NMR reveals intramembrane polar networks in a seven-helical transmembrane protein proteorhodopsin. J Am Chem Soc 133(43):17434–17443. https://doi.org/10.1021/ja207137h
Xiang S, Grohe K, Rovó P, Vasa SK, Giller K, Becker S, Linser R (2015) Sequential backbone assignment based on dipolar amide-to-amide correlation experiments. J Biomol NMR 62(3):303–11. https://doi.org/10.1007/s10858-015-9945-4
Zhou DH, Rienstra CM (2008) High-performance solvent suppression for proton detected solid-state NMR. J Magn Reson 192(1):167–172. https://doi.org/10.1016/j.jmr.2008.01.012
Acknowledgements
We thank the National Facility for High Field NMR in TIFR Hyderabad, where all the experiments were done, Dr. Krishna Rao for technical assistance, and intramural funds at TIFR Hyderabad from the Department of Atomic Energy (DAE). KRM acknowledges TIFR Hyderabad for support under the Young Researcher Programme and Department of Science and Technology (DST), India for funding under the Inspire Faculty Scheme (IFA-CH-150). VA acknowledges support from Science and Engineering Research Board (SERB), DST, India, via Grant No. ECR/2017/001450.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sharma, K., Madhu, P.K., Agarwal, V. et al. Simultaneous recording of intra- and inter-residue linking experiments for backbone assignments in proteins at MAS frequencies higher than 60 kHz. J Biomol NMR 74, 229–237 (2020). https://doi.org/10.1007/s10858-019-00292-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-019-00292-y