
Abstract
We present here 1H-detected triple-resonance H/N/C experiments that incorporate CO–CA and CA–CB out-and-back scalar-transfer blocks optimized for robust resonance assignment in biosolids under ultra-fast magic-angle spinning (MAS). The first experiment, (H)(CO)CA(CO)NH, yields 1H-detected inter-residue correlations, in which we record the chemical shifts of the CA spins in the first indirect dimension while during the scalar-transfer delays the coherences are present only on the longer-lived CO spins. The second experiment, (H)(CA)CB(CA)NH, correlates the side-chain CB chemical shifts with the NH of the same residue. These high sensitivity experiments are demonstrated on both fully-protonated and 100 %-HN back-protonated perdeuterated microcrystalline samples of Acinetobacter phage 205 (AP205) capsids at 60 kHz MAS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The recent advent of probes capable of spinning small-diameter rotors at frequencies of 60 kHz and above (“ultra-fast MAS”) has opened up new possibilities in the structural and dynamic analysis of biomolecular systems (Schanda et al. 2009; Lewandowski et al. 2010; Demers et al. 2011; Huber et al. 2011; Knight et al. 2011, 2012a, b; Lewandowski et al. 2012). Under ultra-fast MAS, the network of 1H dipolar interactions is heavily weakened and, as a consequence, low-power irradiation becomes efficient for heteronuclear decoupling (Ernst et al. 2001, 2003; Kotecha et al. 2007; Laage et al. 2009) and selective cross polarization (CP) (Laage et al. 2008, 2009). In addition, in this spinning regime, the linewidth of the 1H signals is considerably narrowed, and experimental schemes based on direct 1H detection, which require heavy dilution against a perdeuterated background under moderate MAS (Hologne et al. 2006; Linser et al. 2008; Akbey et al. 2010; Linser 2011) become feasible with less stringent dilution constraints, yielding a significant improvement in sensitivity for deuterated molecules with fully-reprotonated amide sites (Zhou and Rienstra 2008, 2007a, b; Knight et al. 2011; Lewandowski et al. 2011) or even for fully protonated samples (Zhou 2007a, b; Marchetti et al. 2012; Zhou et al. 2012). Cumulatively, these factors open the way to the analysis of more complex biological solids that may be available in very limited amounts.
The establishment of sequential connectivities between signals from the protein backbone is the essential step of resonance assignment, which in turn is key to any structural and dynamic characterization by NMR. In the solid-state, this is typically achieved under MAS by pulse sequences that make use of homo- and hetero-nuclear dipolar couplings between the 15N- and 13C- spins of the protein backbone (Luca et al. 2003). However, the performance of many of the existing dipolar-based transfers deteriorates rapidly with increasing MAS frequencies. Most of these schemes have a relatively low efficiency at MAS rates higher than 20–25 kHz, and, with some notable exceptions (Vijayan et al. 2009), they are not viable for resonance assignment at 60 kHz. The development of efficient pulse schemes for resonance assignment in the ultra-fast spinning regime is therefore extremely important.
At high spinning frequencies, sufficiently long-lived 13C and 15N coherences can be achieved without any sample heating under low-power decoupling (Kotecha et al. 2007; Laage et al. 2009), and solution-state NMR techniques become viable, allowing the acquisition of sequential connectivities based on efficient scalar transfer schemes (Bertini et al. 2011; Webber et al. 2012).
Notably, we have previously incorporated J-based 13C–13C transfers into 3D 1H–13C–15N experiments, so to record 1H-detected (H)CO(CA)NH and (H)CA(CO)NH correlations. This was shown to enable the resonance assignment of deuterated and protonated proteins with molecular weights in the 10–16 kDa range (Knight et al. 2011; Marchetti et al. 2012). As compared to the situation in solution-state NMR, however, the efficiency of through-bond transfers is significantly reduced, since in fully-protonated solid samples the transverse coherence lifetimes of the aliphatic carbons do not largely exceed the delay necessary to evolve the scalar coupling between the pair of bonded 13C spins (2τ = 1/(2J CC) = 9.4 ms for a 1 J CACO = 55 Hz and 2τ = 1/(2J CC) = 14.3 ms for a 1 J CACB = 35 Hz) (Lesage et al. 1999; De Paepe et al. 2003). This is because in solid samples the coherence decay is dominated by the coherent contribution from the extended network of 1H–1H dipolar interactions, which is averaged out in solution. Thus while in rigid solids the CO coherences are usually long lived, with T 2 ′ decay time constants of about 40–50 ms, even in fully protonated proteins and at high magnetic fields (Bertini et al. 2011), this is notably not the case for the CA spins, for which the coherence lifetimes are on the order of 20 ms for deuterated, 100 %-HN back-exchanged microcrystalline systems (Knight et al. 2011), and typically not longer than 10–15 ms for protonated samples (Webber et al. 2012). This impacts the performance of both the (H)CA(CO)NH and (H)CO(CA)NH experiments. Moreover, their efficiency is further reduced in the presence of a third coupled spin CB, because the CA → CO transfer may no longer be selective, since some of the CA coherences can leak to the CB spins in cases where the CB resonances lie within or close to the region of CA resonances. In such cases (e.g. threonine, serine or leucine residues), the CA selective pulse also flips the CB spin states, with the result that the CA–CB coupling is not refocused.
Here we show that these drawbacks are overcome with the use of a variant of these experiments incorporating an out-and-back S → I → S homonuclear J-based coherence transfer step. Such blocks are widely employed in multidimensional NMR experiments for the structure determination of proteins in solution (Sattler et al. 1999), both for heteronuclear (H → N and H → C) and homonuclear (CA → CO, CA → CB) transfers, to avoid the evolution of fast-decaying transverse magnetization during the spin echo time periods of the coherence transfer. Similar schemes have been implemented in the solid-state, in extensively deuterated proteins, where the out-and-back experiments are essential to record sequential information when only a small percentage of the amide sites are protonated (Linser et al. 2008; Schanda et al. 2009; Linser 2011).
We show here that the implementation of a homonuclear (CO)CA(CO) J-based coherence transfer block in the (H)CA(CO)NH experiment yields the (H)(CO)CA(CO)NH pulse sequence that records the shifts of CA spins while the transverse magnetization during the spin-echo periods is present only on the CO spins, thus yielding inter-residue correlations with enhanced sensitivity. In addition, we show that a modification of this scheme, the (H)(CA)CB(CA)NH experiment, lends itself to obtaining an additional correlation, this time involving the CB spins.
These new experiments are demonstrated on microcrystalline samples of both fully-protonated and perdeuterated, 100 %-HN back-protonated icosahedral capsids of Acinetobacter phage 205 (AP205) (mol. weight 2.5 MDa, UniProt code: Q9AZ42) under ultra-fast MAS. These particles are composed of 90 subunits, each subunit being a homodimer of a 130-residue nucleoprotein (Klovins et al. 2002).
Experimental section
Sample preparation
The [15N, 13C]-labeled recombinant AP205 coat protein was expressed in E.coli using a modified pETDuet vector (Novagen). Bacteria were grown in a minimal medium enriched with 13C-glucose (2 g/L) and 15N-labeled ammonium chloride (1 g/L) until they reached OD600 = 0.7. IPTG was added to 1 mM final concentration and cells were grown for 4 more hours before being centrifuged and frozen. Lysis buffer (40 mM tris–HCl, pH 8.0, 300 mM NaCl, 1 mg/mL lysozyme, 10 ug/mL DNase, 10 mM MgCl2,) was added (3 mL per gram of cells), and cells were further lysed by sonification. The resulting solution was centrifuged, and the supernatant was loaded on a Sepharose CL-4B (GE Healthcare) column. The resulting fractions containing the capsids were further purified by Fractogel (Merck) ion-exchange column. For crystallization, the hanging drop method was used, and 5 uL of proteins were mixed with 5 uL of precipitant (0.1 M HEPES at pH 7.5, 0.1 M NaCl and 10 % PEG w/v 4,000). The [2H, 15N, 13C]-labeled recombinant AP205 coat protein was expressed similarly, but in 100 % D2O medium using 2H,13C-glucose. For proton exchange of the amide deuterons, the capsids were soaked for 1 h in 2 M guanidinium hydrochloride at room temperature, followed by dialysis against 10 mM HEPES pH 7.5. The purification steps and crystallization conditions were the same as for the doubly-labeled capsids.
NMR experiments
Experiments were carried out on a 800 MHz Bruker Avance III spectrometer using a triple-resonance (1H/13C/15N) 1.3 mm probe. The MAS frequency was 60 kHz and the temperature was regulated to 230 K (corresponding to an approximate sample temperature of 291 K as estimated using the water resonance chemical shift) (Lesage et al. 2008). The pulse sequences presented are given in Figs. 2 and 4. Selective CP (Laage et al. 2008) from 1H to 13C (either CA or CO) was performed using a linear ramp (Hediger et al. 1993; Metz et al. 1994) (90–100 %) with a maximum radio-frequency (RF) amplitude of 100 kHz on 1H and with 20 kHz on 13C for a contact time of 1.5 ms (CA) or 7 ms (CO). Selective CP from 13CO to 15N was performed using a tangential ramp (95–105 % (Hediger et al. 1994)) with a mean radio-frequency (RF) amplitude of 25 kHz on 15N and with 35 kHz on 13C for a contact time of 8 ms. Finally, back CP from 15N to 1H was performed using a linear ramp (100–70 %) with a maximum radio-frequency (RF) amplitude of 100 kHz on 1H and with 20 kHz on 15N for a contact time of 400 μs. Swept low-power two-pulse phase modulated (slTPPM) decoupling (Lewandowski et al. 2010) was applied during all periods of free evolution and acquisition with a 1H RF of 14.3 kHz and a pulse length of 33.3 μs. 1H, 13C and 15N 90°-pulse lengths of 2.5 μs (100 kHz), 3.6 μs (70 kHz) and 5 μs (50 kHz) were used, respectively, along with CO/CA selective Q3 (Emsley and Bodenhausen 1990) (4,000 points) inversion pulses of 700 and 350 μs for the (H)CA(CO)NH and (H)(CO)CA(CO)NH, respectively. An aliphatic-selective Q3 pulse of 200 μs was used in the (H)(CA)CB(CA)NH experiment. In all these experiments, the half-echo delays τ and τ′ were set in both echoes to 4.7 and 7.2 ms for the CA → CO and CA → CB transfers respectively. For the 3D experiments presented in Fig. 2, a total of 34 and 54 points were acquired in t 1 and t 2 , respectively, with 128 scans each. Maximum acquisition times of 6 ms in t 1 , 3.1 ms in t 2 and 20 ms in t 3 and a recycle delay of 1 s were used giving a total experimental time of 65.3 h. For the 3D (H)CANH experiment presented in Fig. 3 (pulse sequence shown in Online Resource Fig. 1), a total of 34 and 60 points were acquired in t 1 and t 2 , respectively, with 48 scans each. Maximum acquisition times of 6 ms in t 1 , 3.5 ms in t 2 and 20 ms in t 3 and a recycle delay of 1 s were used giving a total experimental time of 27.2 h. Finally, for the 3D (H)(CA)CB(CA)NH experiment presented in Fig. 4, a total of 32 and 112 points were acquired in t 1 and t 2 , respectively, with 64 scans each. Maximum acquisition times of 4.8 ms in t 1 , 3.7 ms in t 2 and 20 ms in t 3 and a recycle delay of 1 s were used giving a total experimental time of 63.7 h. In all these 3D experiments, quadrature detection in both indirect dimensions was obtained using States-TPPI (Marion et al. 1989).
Results and discussion
“I → S” versus “S → I → S” homonuclear J-coupling coherence transfers
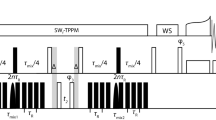
Figure 1 presents the pulse sequences and schematic transfer efficiencies for the two archetypical versions of a through-bond I-S single-quantum (SQ) correlation experiments. The first scheme (“full-forward-transfer” (I → S) block, Fig. 1a) requires initial magnetization on the I spin only, whose chemical shift is immediately recorded in t 1. Anti-phase coherences 2I y S z are created, (“forward”) transferred to spin S (2I z S y ), refocused during the second echo of the sequence, and detected at ΩS in t 2. In the second scheme (“out-and-back transfer” (S → I → S), Fig. 1b), the magnetization is on the S-spin at the beginning of the scalar transfer block. In-phase S-spin coherences are converted (“out”) to anti-phase 2S z I y by a first homonuclear J-coupling coherence transfer period. This allows recording the I spin chemical shift during the indirect acquisition, and the I spin anti-phase at the end of t 1 is converted (“back”) to in-phase S spin coherences, and detected at ΩS in t 2.

Pulse schemes for a “full-forward transfer” I → S and b “out-and-back” S → I → S homonuclear J-based correlations between two spins I and S. The operator terms present at the beginning and at the end of each echo of the sequences are indicated. The filled circle indicates the spin with the initial coherences. c Simulated build up curves for the I-spin in-phase magnetization during the echo delay τ. Bulk T 2 ′ values measured for 13CA and 13CO (spins I and S respectively) at 60 kHz MAS under low-power (15 kHz) decoupling in fully-protonated (left) and perdeuterated 100 %-HN (right) AP205 capsids (T 2′(I) = 8.6 and 25 ms (CA); T 2′(S) = 25.5 and 40 ms (CO), for fully-protonated and perdeuterated, respectively) were used for these calculations. The delay τ’ was set to 4.7 ms, and the coupling constants J IS and J IK used were 53 (J CO–CA) and 35 (J CA–CB) Hz, respectively
Figure 1c shows the calculated intensity buildups of the I-S cross-peaks for each version of the J-correlation experiments. The determining factor for overall sensitivity is the coherence lifetime T 2′. Coherences are scaled during τ and τ′ according to the transverse dephasing times of both spins I and S (i.e. exp(−2τ/T 2′I −2τ′/T 2′S)) for the I → S scheme, but only the coherence lifetime of spin S determines the efficiency in the “out-and-back” S → I → S variant (exp(−(2τ+2τ′)/T 2′S)). The S → I → S experiment is thus more efficient if one of the spins features a particularly long T 2′. The relative efficiencies of the two schemes are estimated here using the experimentally determined T 2′ values for the CO and CA resonances of fully-protonated and deuterated AP205 at ultra-fast MAS. As rationalized above, we clearly see that the S → I → S version gains significantly in sensitivity over the full-forward-transfer I → S scheme due to the absence of fast transverse relaxation on the CA spin. In this experiment, the overall theoretical transfer efficiency is close to 50 % of the initial magnetization for the protonated sample, and close to 60 % in the case of the perdeuterated 100 %-HN sample.
In addition, the relative performance of the two experiments is affected differently in the presence of a third spin K coupled to the first spin I with a coupling constant J IK . In this case, in the “full-forward-transfer” scheme, a portion of the initial magnetization is transferred into K-spin antiphase coherences, and does not contribute to the intensity of the observed I–S cross-signal. On the contrary, the S → I → S variant does not generate any I-spin coherences, and its efficiency is thus independent of the presence of additional I spin coupling partners.
CA–CO transfers in triple-resonance experiments
Figure 2 shows the implementation of these two versions of the homonuclear 13C–13C J-based coherence transfer in the context of protein backbone sequential assignment based on 1H-detection experiments. Here spin I = CA and spin S = CO.

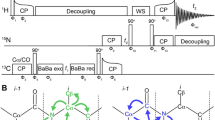
Pulse sequences for the proton-detected CA i Ni+1H i+1 sequential assignment experiments, and proton-detected triple-resonance spectra recorded on protonated and perdeuterated 100 %-HN (15N,13C)-labelled capsids of AP205 at 60 kHz MAS under low-power decoupling. Pulse sequences for a (H)CA(CO)NH and b (H)(CO)CA(CO)NH. Narrow and broad black rectangles indicate π/2 and π pulses, respectively, and bell shapes represent band-selective π pulses. More details are given in the experimental section. In scheme a, as CO and CA selective pulses cannot be applied simultaneously in the first half of the homonuclear J-based transfer block (pink box), an extra pulse needs to be applied on CO to compensate for the chemical shift evolution. The refocusing echo of scheme (a) and both echoes in scheme (b) (purple boxes) use broadband π-pulses since only CO coherences are present, therefore only the CO-CA coupling needs to be considered. c–j 1D proton-detected triple-resonance spectra recorded on protonated (c–f) and perdeuterated 100 %-HN (g–j) capsids. c/g (H)CANH; d/h (H)CONH; e/i (H)CA(CO)NH; f/j (H)(CO)CA(CO)NH. Pulse sequences for (H)CANH and (H)CONH are presented in Online Resource Fig S1. k–l 2D 15N–13C projections of the 3D proton-detected (H)CA(CO)NH (k) and (H)(CO)CA(CO)NH (l) spectra of fully-protonated (15N,13C)-labelled capsids. Resonances are labeled according to the residue of their CA spin. Traces along the 13C dimension are shown to illustrate the gain in sensitivity
In the CA(CO) variant (Fig. 2a), the CA chemical shift is encoded during the first indirect dimension after a 1H–13CA selective CP, then coherences are transferred to CO, and from there to 15N by a 13CO–15N CP. In the version of the experiment incorporating the out-and-back (CO)CA(CO) J-based coherence transfer block (Fig. 2b), the magnetization is first transferred from 1H to CO instead, then the in-phase CO coherences are converted to anti-phase 2CA y CO z by a first J-based coherence transfer echo period. This allows recording the CA chemical shift during the indirect acquisition, and the CA anti-phase at the end of t 1 is converted back to in-phase CO coherences.
Because CA is coupled to both CO (J CA–CO = 55 Hz) and CB (J CA–CB = 35 Hz), a key requirement in the first version of the experiment is to avoid the generation of coherences that are anti-phase with respect to the CA–CB J coupling. This relies on the use of π-pulses which are selective enough to refocus the CA spins but not the CB spins. This is impractical, however, since the CA and CB of some residue types resonate outside the characteristic CA and CB chemical shift ranges (45–65 ppm and <45 ppm, respectively), notably Gly CA (around 45 ppm) and Thr/Ser CB (60–70 ppm). Typically, either a very long CA selective pulse can be used, which cuts out the Gly signals, or a shorter, less selective pulse that refocuses all the CAs, but reintroduces the J coupling to some of the less shifted CB resonances.
This is not an issue in the scheme incorporating the out-and-back (CO)CA(CO) step, where coherences during the echo delays of the scalar transfer units are always on the CO spins experiencing a single large J-coupling to the CA spins. In this case, the difficulties associated with employing selective pulses during the spin echo of the transfer blocks are avoided, since non-selective pulses can be applied.
The overall efficiency offered by the out-and-back (CO)CA(CO) step is expected to be superior. On one side, the transfer between the CO and CA is achieved with the coherence present on the longer-lived CO spin during both spin echo periods. With a τ = 4.7 ms and CO and CA coherence lifetimes of 25.5 and 8.6 ms, respectively, the (CO)CA(CO) scheme is expected to yield a sensitivity improvement of a factor of 1.4 (Fig. 1c).
On the other side, a non-negligible part of the magnetization is inevitably lost in the CA(CO) transfer scheme during the three selective pulses of 1 ms each applied during the first scalar transfer spin echo block. This accounts for a further reduction in the intensity.
Figure 2c–f compares the relative efficiency of the two types of experiments on fully-protonated 13C,15N-labelled AP205 capsids at 800 MHz under 60 kHz MAS. Despite the similar efficiencies of the initial H → CA and H → CO transfers, as well as the final CA → N and CO → N transfers (as can be seen by comparing the first increments of an (H)CANH and (H)CONH spectra, Fig. 2c, d), the insertion of the (CO)CA(CO) step yields a 40–50 % higher efficiency in bulk as compared to the direct transfer experiment. This is shown by comparing the S/N ratios of the first increment of the respective 3D spectra (Fig. 2e, f).
Figure 2k and l show the N–CA projections of two 3D (H)CA(CO)NH and (H)(CO)CA(CO)NH datasets. Most of the resonances feature an average 50 % higher intensity in the (H)(CO)CA(CO)NH experiment. Moreover, in the CA(CO) variant, we see that 20 signals out of 100 are extremely weak, notably from all the glycines, which are not touched by the CA-selective refocusing pulse, and from the threonines (e.g. T92 and T106), serines (e.g. S84 and S110) and leucines (e.g. L24 and L115), whose CBs resonate within the CA-selective pulse bandwidth. A more selective CA pulse allows one to partially recover these signals, but at the expense of the overall sensitivity (see Online Resource, Figure 2). On the contrary, all these signals are unambiguously observed in the (H)(CO)CA(CO)NH experiment.
Note that the advantages of this approach, here demonstrated on a fully-protonated sample, are also pertinent in the case of perdeuterated samples fully reprotonated at the amide sites, as illustrated in Fig. 2g–j. In these samples, not only the CO spins have longer coherence lifetimes than CA spins but, additionally, the efficiency of the initial H–CO CP transfer step is higher than the corresponding transfer from H to CA (Fig. 2 g, h). This further justifies the use of the out-and-back transfer with respect to the protonated case, since this implementation of the sequence only involves CP transfer steps to and from CO spins. Moreover, also in this case the use of non-selective pulses allows the observation of all inter-residue resonances, including those from serine, threonine or glycine (data not shown).
Overall, this new experiment allows the acquisition of inter-residue correlation with a two-fold reduction of the experimental time with respect to the full-transfer experiment, regardless of the residue type. The availability of sensitive inter-residue CA–N–H maps provides key information for constructing the sequential resonance assignment along the protein backbone. Figure 3 illustrates a stretch of the walk along the sequence obtained by aligning strips from intra-residue (H)CANH and inter-residue (H)(CO)CA(CO)NH.

2D 15N–13C projections of the 3D proton-detected (H)CANH (a) and (H)(CO)CA(CO)NH (b) spectra necessary for sequential assignment of protonated (15N,13C)-labelled protonated capsids of AP205 recorded on a 800 MHz spectrometer at 60 kHz MAS under low-power decoupling. An illustrative part of the sequential walk is shown in (c)
We note that, using the same notation, the equivalent out-and-back experiments in solution-state NMR can be described as (HNCO)CA(CO)NH and (HNCA)CB(CA)NH, where one-bond J-couplings are used to facilitate both the hetero- and homonuclear coherence transfers. For these sequences, the term ‘out-and-back’ means that the coherence is transferred along the chain of nuclei from the amide 1H to the 13C whose shift is monitored during t 1, and then returned to the same 1H along the same path. In the solid state, we are able to use selective CP to polarize either the 13CO or 13CA in the first transfer step of the ‘out’ part of the sequence without the need to involve the 15N in an intermediate step. So although the ‘out’ transfer follows a shorter path than the ‘back’ transfer, the new solid-state sequences have a similar structure to their solution-state counterparts, and so we use the term ‘out-and-back’ for both.
CB–CA transfers in triple-resonance experiments
The same approach can be used to acquire experiments comprising the chemical shift evolution of side-chain nuclei. For example, an evident extension of the scheme presented in the previous section is the (H)(CA)CB(CA)NH experiment, which allows one to record a correlation between the backbone amide N and H, and the side-chain CB.
In this experiment (Fig. 4a), the magnetization is first transferred from 1H to CA. The generated in-phase CA coherences are converted to anti-phase 2CB y CA z by a first J-based coherence transfer echo period. This allows recording the CB chemical shift during the indirect acquisition, and the CB anti-phase at the end of t 1 is converted back to in-phase CA coherences. Simulated build up curves for CB in-phase magnetization during the echo delay τ in a (H)(CA)CB(CA)NH experiment are reported in the Online Resource Figure 3.

a Pulse sequence for the proton-detected CB i NiH i correlation experiment. Narrow and broad black rectangles indicate π/2 and π-pulses, respectively, and bell shapes represent aliphatic-selective inversion pulses. b Overlay of the 2D 15N–13C projections of the 3D proton-detected (H)CANH (green) and (H)(CA)CB(CA)NH (purple) spectra of protonated (15N, 13C)-labelled capsids of AP205 recorded on a 800 MHz spectrometer at 60 kHz MAS under low-power decoupling
Compared to the CA–CO transfer experiment, the π-pulses applied during the two echo periods simply need to affect both CA and CB for the evolution of the J CACB (35 Hz) while leaving the CO spins untouched to refocus the J CACO. In practice this only requires to apply a short selective pulse covering the whole aliphatic region (about 80 ppm).
An overlay of the 2D projections from intra-residue (H)CANH (green) and (H)(CA)CB(CA)NH (purple) 3D experiments is shown in Fig. 4b for the fully-protonated capsids of AP205. About 70 fully-resolved H–N–CB cross-signals could be observed in an overall experimental time of 3 days.
Notably, this solution is also extremely pertinent for resonance assignment of perdeuterated samples, where the efficiency of the alternative dipolar-based schemes (Zhou et al. 2012) is not only weakened at ultra-fast MAS, but also involves an inefficient H → CB step. Here, with the (H)(CA)CB(CA)NH experiment we were able to detect all of the 100 expected CB resonances in the perdeuterated 100 %-HN sample of AP205 capsids in 2.5 days of experimental time (Online Resource, Figure 4).
The availability of sensitive intra-residue CB–N–H correlations combined with intra-residue CA–N–H correlations is crucial, as it provides key information for determining residue types, and for confirming the assignment made on the basis of backbone resonances only.
Conclusions
The availability of sensitive inter-residue CA–N–H maps is critical for sequence-specific protein backbone resonance assignment. The (H)(CO)CA(CO)NH scheme, which makes use of an out-and-back (CO)CA(CO) homonuclear J-based coherence transfer, allows one to record 3D inter-residue through-bond correlation spectra in fully-protonated and 100 %-HN perdeuterated biosolids with high sensitivity under ultra-fast MAS. The experiment proposed here encodes the chemical shift of the CA spin while exploiting the long coherence lifetime of the neighboring CO spin. As compared to full-transfer schemes, CA correlations are observed regardless of the residue type and with a two-fold reduction in the experimental time. An intra-residue (H)(CA)CB(CA)NH implementation of this scheme is introduced, and provides access to CB chemical shifts, which are key information for robust protein assignment.
These experiments are expected to become essential tools for the study of crystalline and non-crystalline biomolecules by NMR in the solid state.
References
Akbey U, Lange S, Franks WT, Linser R, Rehbein K, Diehl A, van Rossum BJ, Reif B, Oschkinat H (2010) Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR 46:67–73
Bertini I, Emsley L, Felli IC, Laage S, Lesage A, Lewandowski JR, Marchetti A, Pierattelli R, Pintacuda G (2011) High-resolution and sensitivity through-bond correlations in ultra-fast magic angle spinning (MAS) solid-state NMR. Chem Sci 2:345–348
De Paepe G, Giraud N, Lesage A, Hodgkinson P, Böckmann A, Emsley L (2003) Transverse dephasing optimized solid-state NMR spectroscopy. J Am Chem Soc 125:13938–13939
Demers JP, Chevelkov V, Lange A (2011) Progress in correlation spectroscopy at ultra-fast magic-angle spinning: basic building blocks and complex experiments for the study of protein structure and dynamics. Solid State Nucl Magn Reson 40:101–113
Emsley L, Bodenhausen G (1990) Gaussian pulse cascades—new analytical functions for rectangular selective inversion and in-phase excitation in NMR. Chem Phys Lett 165:469–476
Ernst M, Samoson A, Meier BH (2001) Low-power decoupling in fast magic-angle spinning NMR. Chem Phys Lett 348:293–302
Ernst M, Samoson A, Meier BH (2003) Low-power XiX decoupling in MAS NMR experiments. J Magn Reson 163:332–339
Hediger S, Meier BH, Ernst RR (1993) Cross-polarization under fast magic-angle sample-spinning using amplitude-modulated spin-lock sequences. Chem Phys Lett 213:627–635
Hediger S, Meier BH, Kurur ND, Bodenhausen G, Ernst RR (1994) NMR cross-polarization by adiabatic passage through the hartmann-hahn condition (APHH). Chem Phys Lett 223:283–288
Hologne M, Chevelkov V, Reif B (2006) Deuterated peptides and proteins in MAS solid-state NMR. Prog Nucl Magn Reson Spectrosc 48:211–232
Huber M, Hiller S, Schanda P, Ernst M, Böckmann A, Verel R, Meier BH (2011) A proton-detected 4D solid-state NMR experiment for protein structure determination. ChemPhysChem 12:915–918
Klovins J, Overbeek GP, van den Worm SH, Ackermann HW, van Duin J (2002) Nucleotide sequence of a ssRNA phage from Acinetobacter: kinship to coliphages. J Gen Virol 83:1523–1533
Knight MJ, Webber AL, Pell AJ, Guerry P, Barbet-Massin E, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Emsley L, Lesage A, Herrmann T, Pintacuda G (2011) Fast resonance assignment and fold determination of human superoxide dismutase by high-resolution proton-detected solid-state MAS NMR spectroscopy. Angew Chem Int Ed 50:11697–11701
Knight MJ, Felli IC, Pierattelli R, Bertini I, Emsley L, Herrmann T, Pintacuda G (2012a) Rapid measurement of pseudocontact shifts in metalloproteins by proton-detected solid-state NMR spectroscopy. J Am Chem Soc 134:14730–14733
Knight MJ, Pell AJ, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Herrmann T, Emsley L, Pintacuda G (2012b) Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc Natl Acad Sci 109:11095–11100
Kotecha M, Wickramasinghe NP, Ishii Y (2007) Efficient low-power heteronuclear decoupling in 13C high-resolution solid-state NMR under fast magic angle spinning. Magn Reson Chem 45:S221–S230
Laage S, Marchetti A, Sein J, Pierattelli R, Sass HJ, Grzesiek S, Lesage A, Pintacuda G, Emsley L (2008) Band-selective 1H–13C cross-polarization in fast magic angle spinning solid-state NMR spectroscopy. J Am Chem Soc 130:17216–17217
Laage S, Lesage A, Emsley L, Bertini I, Felli IC, Pierattelli R, Pintacuda G (2009) Transverse-dephasing optimized homonuclear J-decoupling in solid-state NMR Spectroscopy of uniformly 13C-labeled proteins. J Am Chem Soc 131:10816–10817
Lange A, Scholz I, Manolikas T, Ernst M, Meier BH (2009) Low-power cross polarization in fast magic-angle spinning NMR experiments. Chem Phys Lett 468:100–105
Lesage A, Bardet M, Emsley L (1999) Through-bond carbon–carbon connectivities in disordered solids by NMR. J Am Chem Soc 121:10987–10993
Lesage A, Gardiennet C, Loquet A, Verel R, Pintacuda G, Emsley L, Meier BH, Böckmann A (2008) Polarization transfer over the water-protein interface in solids. Angew Chem Int Ed 47:5851–5854
Lewandowski JR, Sein J, Sass HJ, Grzesiek S, Blackledge M, Emsley L (2010) Measurement of site-specific 13C spin-lattice relaxation in a crystalline protein. J Am Chem Soc 132:8252–8255
Lewandowski JR, Dumez JN, Akbey U, Lange S, Emsley L, Oschkinat H (2011) Enhanced resolution and coherence lifetimes in the solid-state NMR spectroscopy of perdeuterated proteins under ultrafast magic-angle spinning. J Phys Chem Lett 2:2205–2211
Lewandowski JR, Sass HJ, Grzesiek S, Blackledge M, Emsley L (2012) Site-specific measurement of slow motions in proteins. J Am Chem Soc 133:16762–16765
Linser R (2011) Side-chain to backbone correlations from solid-state NMR of perdeuterated proteins through combined excitation and long-range magnetization transfers. J Biomol NMR 51:221–226
Linser R, Fink U, Reif B (2008) Proton-detected scalar coupling based assignment strategies in MAS solid-state NMR spectroscopy applied to perdeuterated proteins. J Magn Reson 193:89–93
Luca S, Heise H, Baldus M (2003) High-resolution solid-state NMR applied to polypeptides and membrane proteins. Acc Chem Res 36:858–865
Marchetti A, Jehle S, Felletti M, Knight MJ, Wang Y, Xu ZQ, Park AY, Otting G, Lesage A, Emsley L, Dixon NE, Pintacuda G (2012) Backbone assignment of fully protonated solid proteins by 1H detection and ultrafast magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed 51:10756–10759
Marion D, Ikura M, Tschudin R, Bax A (1989) Rapid recording of 2D NMR-spectra without phase cycling—application to the study of hydrogen-exchange in proteins. J Magn Reson 85:393–399
Metz G, Wu XL, Smith SO (1994) Ramped-amplitude cross-polarization in magic-angle-spinning NMR. J Magn Res A 110:219–227
Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc 34:93–158
Schanda P, Huber M, Verel R, Ernst M, Meier BH (2009) Direct detection of 3hJNC’ hydrogen-bond scalar couplings in proteins by solid-state NMR spectroscopy. Angew Chem Int Ed 48:9322–9325
Vijayan V, Demers JP, Biernat J, Mandelkow E, Becker S, Lange A (2009) Low-power solid-state NMR experiments for resonance assignment under fast magic-angle spinning. ChemPhysChem 10:2205–2208
Webber AL, Pell AJ, Barbet-Massin E, Knight MJ, Bertini I, Felli IC, Pierattelli R, Emsley L, Lesage A, Pintacuda G (2012) Combination of DQ and ZQ coherences for sensitive through-bond NMR correlation experiments in biosolids under ultra-fast MAS. ChemPhysChem 13:2405–2411
Zhou DH, Rienstra CM (2008) Rapid analysis of organic compounds by proton-detected heteronuclear correlation NMR spectroscopy with 40 kHz magic-angle spinning. Angew Chem Int Ed Engl 47:7328–7331
Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM (2007a) Proton-detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning. J Am Chem Soc 129:11791–11801
Zhou DH, Shea JJ, Nieuwkoop AJ, Franks WT, Wylie BJ, Mullen C, Sandoz D, Rienstra CM (2007b) Solid-state protein-structure determination with proton-detected triple-resonance 3D magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed 46:8380–8383
Zhou DH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, Shah GJ, Brea EJ, Lemkau LR, Rienstra CM (2012) Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR 54:291–305
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Barbet-Massin, E., Pell, A.J., Jaudzems, K. et al. Out-and-back 13C–13C scalar transfers in protein resonance assignment by proton-detected solid-state NMR under ultra-fast MAS. J Biomol NMR 56, 379–386 (2013). https://doi.org/10.1007/s10858-013-9757-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-013-9757-3