
Abstract
Electrophoretic deposition (EPD) is a facile and feasible technique to prepare functional nanocomposite coatings for application in orthopedic-related implants. In this work, a ternary graphene oxide–chitosan–hydroxyapatite (GO–CS–HA) composite coating on Ti substrate was successfully fabricated by EPD. Coating microstructure and morphologies were investigated by scanning electron microscopy, contact angle test, Raman spectroscopy, Fourier transform infrared spectroscopy and thermogravimetric analysis. It was found GO–CS surface were uniformly decorated by HA nanoparticles. The potentiodynamic polarization test in simulated body fluid indicated that the GO–CS–HA coatings could provide effective protection of Ti substrate from corrosion. This ternary composite coating also exhibited good biocompatibility during incubation with MG63 cells. In addition, the nanocomposite coatings could decrease the attachment of Staphylococcus aureus.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Electrophoretic deposition (EPD) is a colloidal coating technique that uses electric filed to move charged particles towards working electrode and deposit [1, 2]. The interest in EPD is not only from its high versatility but also from its cost effectiveness. It is a simple process with capability of fabricating multilayer and functionally graded coatings. Nowadays, EPD is gaining increased attention in thin films for biomedical applications. The initial usage of EPD in biomaterials field was developing hydroxyapatite (HA) coatings on metallic orthopedic implants, where scientists aimed to enhance the bioactivity of metal surfaces and thus promotes the integration of the implants with surrounding tissues. As the key component of natural bone, HA shows excellent biocompatibility and osteoconductivity [3], and has been widely explored as functional coatings for orthopedic implants. It is convenient to control deposited HA stoichiometry and thickness in a wide range through EPD by changing processing parameters, for example, time, voltage and concentration. However, pure HA coatings are usually subjected to shrink and crack during sintering at high temperature [4]. To eliminate the problems related to sintering and further improve the bioactivity and functionalities of HA, preparation of biomimetic organic-HA nanocomposite coatings has been a hot research topic, such as HA–chitosan [5], HA–hyaluronic acid [4, 6], HA–alginic acid [7], HA–chitosan–heparin [8] and etc. As a derivative of chitin, chitosan (CS) is a natural linear cationic polysaccharide with superior biocompatibility, biodegradability and antibacterial property. CS can promote the expression of extracellular matrix proteins in human osteoblasts and chondrocytes due to its structural similarity to glycosaminoglycans. It can also stimulate the differentiation of osteoprogenitor cells, and thus facilitate bone repair and regeneration [9]. The combination of HA nanoparticles and CS could display a favorable synergetic bioactivity in terms of osteoinductivity and osteoconductivity [10, 11]. CS also owns excellent film forming properties and the CS–HA composite coatings prepared through EPD have shown good adhesion to the substrate [5].
Graphene oxide (GO), a novel two-dimensional nanomaterial, has attracted enormous research interests due to its fascinating properties: high specific surface area, excellent mechanical properties, good dispensability in both water and some organic solvents, desirable biocompatibility and antibacterial properties [12]. Consequently, GO could have a wide variety of promising application potentials in biomedical engineering area, such as biosensor, drug or gene delivery system, hydrogel and tissue engineering [13–18]. The incorporation of graphene-based materials with HA composites has been widely employed by many fabrication techniques, notably, spark plasma sintering (SPS), hydrothermal treatment, chemical precipitation method, electrodeposition, and wet-mixing process. With graphene serving as effective enforcement fillers, all composite films exhibited improved mechanical performance compared to pure HA [19–24]. Zhang synthesized graphene/HA composite using SPS and found that 1.0 wt% of graphene in composite could lead to nearly 80 % improvement in fracture toughness [19]. Sintered composites of reduced graphene oxide (rGO) and HA prepared by hydrothermal method demonstrated an increase of elastic modulus and fracture toughness by 86 and 40 % respectively [21]. Correspondingly, various GO-biopolymer nanocomposites have been studied, including GO–CS [25–27], GO–gelatin [28], GO–fibrin [29], GO–cellulose acetate [30], GO–PLGA–collagen [31], GO–silk [32], GO–alginate [33] and so on. Specifically, GO–CS nanocomposite fabricated by self-assembly in aqueous media is of great interest [34]. For instance, Zuo reported the preparation of free standing GO–CS nanocomposite films through amide linkages between GO and CS [35]. Ordikhani developed GO–CS films containing antibiotics through EPD, and the film showed high bactericidal property [24]. Liu successfully utilized glutaraldehyde cross-linked CS–GO composite to absorb Au (III) and Pd (III) in aqueous solution [36].
In this work, we have taken the processing advantages of EPD, the individual and synergistic biological functions of GO, CS and HA into consideration, and prepared GO–CS–HA nanocomposite coatings on Ti substrate. The goal is to investigate the feasibility of EPD technique, and to evaluate the surface morphology, corrosion resistance, cytotoxicity and the anti-microbial adhesion ability of the coated samples. Hopefully, we could provide guidance for GO-composite application as clinical implants coatings.
2 Materials and methods
2.1 Materials
Pristine GO platelets were purchased from Angstron Material Inc. (Dayton Ohio, USA). Chitosan with low molecular weight (with a deacetylation degree of 75–85 %) was provided by Sigma-Aldrich Co. HA nanoparticles with a particle size of 40 nm were obtained from Nanjing Emperor Nano Material Co., Ltd. Commercial pure Ti (grade TA2) sheet with 1 mm thickness was supplied by Xi’an State Metal Materials Development Co., Ltd. Ti samples (10 mm × 10 mm × 1 mm) were polished by SiC paper (400 grits) and ultrasonically cleaned in acetone, alcohol and deionized (DI) water in turn. All other reagents and solvents of analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd, and used as received without further purification unless otherwise noted.
2.2 Electrophoretic deposition of the composites coatings
Briefly, CS solution (6.4 mg/mL) was dissolved in 1 % acetic acid. HA nanoparticles (6 mg/mL) were dispersed in ethanol with stirring for 3 h, and then added into the CS solution. The pristine GO platelets were pre-exfoliated by ultrasonic dispersion in a mixture of ethanol and DI water (80 vol% of ethanol) overnight and stirred for 4 h with a concentration of 0.076 and 0.044 mg/mL, respectively. Subsequently, GO dispersions were slowly added into aforementioned CS–HA mixture, and the resulting mixed suspensions were ultrasonic dispersed for 20 min and then stirred for 3 h before EPD. The final deposition suspensions (80 vol% of ethanol) consisted of 0.64 mg/mL CS, 1.5 mg/mL HA nanoparticles and different concentrations of GO platelets (0.0, 1.0 and 1.7 wt%, respectively.)
The EPD processes were conducted under continuous stirring using 20 V DC power for 3 min with a stainless steel electrode (anode) and a Ti substrate (cathode) 1 cm apart. The coated samples were taken out carefully from the EPD cell and dried horizontally in air at room temperature for 24 h. The resulting coatings were denoted as CS–HA, 1.0 GO–CS–HA and 1.7 GO–CS–HA, respectively based on different GO contents. In addition, samples coated with pure HA by EPD are used as control for in vitro biocompatibility studies and the antibacterial assay. The coating process was carried out in 1.5 mg/mL HA nanoparticle suspension (80 vol% ethanol).
2.3 Characterization
2.3.1 Characterization of deposition suspension
The deposition suspensions with different GO contents were characterized by transmission electron microscope (TEM, H-9000NAR, Hitachi) under 100 kV accelerating voltage. The process of sample preparation was as follows: the colorless suspension diluted with DI water was dropped on the copper mesh surface and then dried at room temperature.
The zeta potentials of the suspensions were evaluated by Zetasizer Nano90 (Malvern Instruments, UK) using dynamic light scattering. The suspensions were ultrasonically treated to ensure its homogeneous distribution before test.
2.3.2 Surface morphology and microstructure analysis
The coating morphology was observed by scanning electron microscope (SEM, AMRY 1-1910FE). The water contact angle was measured by the sessile drop method with an automatic drop shape analysis (SL200B, America). Specimens in triplicate were tested for each composite coating. The microscopic Fourier transform infrared analysis (Micro-FTIR, Thermo Fisher) and Raman spectra (RM-100, Renishaw) were utilized to investigate the functional groups of the coatings. The thermal stability of composite coatings were evaluated by thermogravimetric analysis (TGA, TA instrument, Q50) at a heating rate of 10 °C/min from 20 to 600 °C under a 100 mL N2 per min purge.
2.3.3 Electrochemical tests
The corrosion resistant property of the nanocomposite coatings were evaluated by electrochemical test in simulated body fluid (SBF) using a three-electrode workstation (CHI 650C, China) with a working electrode (the specimen), a Pt counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The preparation procedure of SBF can be found from publication of Kokubo [37]. The composition of the SBF (pH 7.4) was as follows: Na+ 142.0 mM, K+ 5.0 mM, Mg2+ 1.5 mM, Ca2+ 2.5 mM, HCO3 − 4.2 mM, Cl− 147.8 mM, HPO4 2− 1.0 mM and SO4 2− 0.5 mM.
2.3.4 Cell viability
The cytotoxicity of the prepared EPD coating towards MG63 cells was evaluated by a cell counting kit-8 assay (CCK-8, Dojindi). MG63 cells were routinely cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS) and 1 % penicillin/streptomycin in a humidified atmosphere of 5 % CO2 at 37 °C.
Cells with an initial number of 3 × 10−4 cells/well were seeded on the sterile bare Ti samples and EPD HA, CS–HA, 1.7 GO–CS–HA coatings in triplicate and then cultured for 1, 3 and 5 days in 24-well plates. DMEM medium without samples served as negative control and 10 % DMSO in DMEM medium served as positive control. At predetermined time points, CCK-8 solution was added to each well. After 2 h incubation, medium optical density (OD) was measured by a spectrophotometer (Elx-800, bio-Tek instruments) at a 450 nm wavelength. The cell viability was expressed as following:
2.3.5 Alkaline phosphatase (ALP) enzyme activity
To measure alkaline phosphatase (ALP) activity, MG63 cells were cultured on the specimen surfaces for 7 days with an initial number of 3 × 104 cells/well. After the culture period, the culture medium was removed and 200 μL of 1 % Triton X-100 was added to the well and treated for 1 h to obtain cell lysates. ALP activity was determined by measuring the light absorbance at 520 nm. ALP in cell lysates could hydrolyze phenylphosphate to phenol and phosphate; phenol could react with 4-aminoantipyrine in presence of potassium ferricyanide in basic solution to form a red-colored complex whose absorbance is directly proportional to the ALP activity in the samples. For normalization, the total protein content was measured by a Bicinchoninic Acid (BCA) protein assay kit (Prod # 23225, Thermo Scientific, USA).
2.3.6 Apatite mineralization
To test biomineralization ability of the coatings in vitro, the pristine Ti, HA, CS–HA and 1.7 GO–CS–HA coatings were soaked in 1.5 SBF at 37 °C for 5 days with periodical solution replacement. After the treatment, samples were taken out, dried, and examined for apatite formation using SEM. The 1.5 SBF has 1.5× ion concentrations than described above in Sect. 2.3.3.
2.3.7 Anti-bacterial adhesion analysis
The anti-bacterial adhesion ability of the coatings was assessed by SEM observation of the gram-positive bacteria Staphylococcus aureus (S. aureus) adhesion on the surface of the specimens. S. aureus cultivated in Luria-Bertani (LB) medium (containing 10 g/L peptone, 5 g/L yeast extract, and 10 g/L sodium chloride) were activated and incubated under vibration with 180 rpm/min at 37 °C. The sterilized specimens were incubated in 24-well tissue plate in 500 μL S. aureus suspension with an initial density of 1 × 105 cells/mL. After co-incubated for 12 h at 37 °C, all sample surfaces were gently rinsed with PBS solution and then fixed in 2.5 % glutaraldehyde solution for 2 h followed by dehydration with a series of graded ethanol solutions for SEM.
3 Results and discussion
3.1 Fabrication of GO–CS–HA coatings by electrophoretic deposition
EPD is usually carried out in a two-electrode cell where the metallic substrate acts as cathode or anode depending on the charge of the deposition particles. In this study, the positive charged GO–CS–HA particles deposited on the titanium substrate acting as the cathode in the electric field with 20 V. The preparation of the suspension and the process of EPD were illustrated in Fig. 1.

Schematic illustration of the preparation process of composite coatings on titanium
3.2 Zeta potentials and dispersion behaviors of the suspensions
Good dispersion and high stability of charged particles are needed to carry out EPD process successfully in the colloids solution. Thus, Zeta potentials of GO–CS–HA suspensions with different GO contents were investigated to check suspension stabilities. A higher Zeta potential is favorable. It was found that 25.8 ± 0.6 mV for CS–HA, 30.1 ± 1.7 mV for 1.0 GO–CS–HA and 36.8 ± 0.8 mV for 1.7 GO–CS–HA. Clearly, dispersion and stability of charged particles were improved by the addition of GO. Zeta potentials of 1.0 GO–CS–HA and 1.7 GO–CS–HA suspensions exceeded the criteria value beyond 30 mV, at which it is recognized as stable colloids suspension [38]. The results also indicated the presence of positively charged particles in the suspensions that is also highly required in this process.
The dispersion uniformity of HA nanoparticles in CS–HA, 1.0 GO–CS–HA and 1.7 GO–CS–HA suspensions were further assessed by TEM observation. Due to their high surface energy, needle-like HA nanoparticles formed a small number of aggregates in CS–HA sample (Fig. 2b). When GO was added into CS–HA suspensions, HA nanoparticles distributed uniformly on the surfaces and edges of lamellar GO–CS matrix (Fig. 2c, d). Thus, GO improved the dispersion of HA nanoparticles, and formed more stable suspension which has been verified by zeta potential analysis.

Zeta potentials (a) and TEM images of the CS–HA (b), 1.0 GO–CS–HA (c), 1.7 GO–CS–HA (d) suspensions, respectively
Due to various colloidal dispersion behaviors, several mechanisms of deposition formation have been established, for example, particle charge neutralization, flocculation by particle accumulation, electrochemical coagulation of particles and electrical double layer distortion and thinning mechanism [2]. Based on results above, a possible mechanism is proposed here to explain the interactions between HA, CS and GO particles and the deposition formation process. Firstly, when HA solution was added into the anionic chitosan solution, the positively charged chitosan molecules attracted HA nanoparticles, and achieved an electrostatic stabilization, therefore, EPD of HA particles was promoted [5]. Secondly, GO sheets are attached with many oxygen-containing hydrophilic functional groups, such as –COOH and –OH [12]. When GO dispersion was added to HA–CS mixture, the negatively charged GO sheets would self-assemble with protonated amine groups in CS molecules. This effect has also been found in Ref. [39] that a stable and homogeneous dispersion of CS–GO could be achieved via GO nanosheets wrapping or attaching to the chitosan macromolecules. Moreover, GO sheets could act as a surfactant as well with an edge-to-center distribution of hydrophilic and hydrophobic domains [40]. Under the influence of an electric field, the positively charged particles moved to the cathode, where the generation of OH− ions led to an increase of the pH value, therefore, amine groups of CS macromolecules became deprotonated [25]. Additionally, the pressure exerted by electric field might further promote the particles accumulation as the flocculation by particle accumulation mechanism and the previous studies suggested [2, 41, 42]. More researches concerning the deposition mechanism are desired to be conducted to shed light on this process. Overall, the co-deposition via polyelectrolyte absorption on the ceramic particles has been achieved in this ternary system of GO–CS–HA by HA bonding to CS and GO.
3.3 Characteristics of the coatings
As shown in the Fig. 3, the composite coatings are rough and compact with only few micro-cracks observed in high magnification image (Fig. 3d). Specifically, the needle-shape HA nanoparticles with approximately 150 nm size were embedded in CS and CS–GO matrices. Comparing with the CS–HA coating, the incorporation of GO resulted in a more coarse morphology. From the high magnification image of 1.7 GO–CS–HA (Fig. 3d) coating, it was found that HA nanoparticles decorated on the edge of some wrinkles which may be caused by GO.

The SEM images of the composite coatings, CS–GO (a), 1.0 CS–GO–HA (b) and 1.7 GO–CS–HA (c) and the corresponding high magnification of 1.7 GO–CS–HA (d), respectively. The insets show the contact angles of the composite surfaces
The contact angles of different coatings were 82.3 ± 1.1°, 76.0 ± 3.2° and 53.3 ± 1.3°, respectively. It is clear that the water contact angle decreased with increasing GO contents, indicating improved wet ability. There were reports also proved GO coatings had high wet ability [43–45]. For instance, Ganesh investigated the effect of GO on the contact angles of GO-polysulfone composite membranes. The result revealed that with GO addition, the membrane became more hydrophilic [44]. Water droplet could be absorbed immediately by GO substrate, but not so quick on PMMA grafted GO [45]. The hydrophilicity of GO can be ascribed to the existence of a large amount of oxygen-functional group. The roughness of its surface could also affect the contact angle due to the increased areas of solid–liquid interface and the effect of sharp edges of rough surfaces [46]. In this study, it was conjectured that the increased GO content and the change of morphology of different coatings co-contributed to the improvement of hydrophilicity of the composite coatings. Moreover, in vitro and in vivo studies have shown that high hydrophilicity is preferred for cell and cell-adhesive proteins adhesion to promote osteoconductivity [47–49].
The compositions of resulting GO–CS–HA composites were investigated by micro FTIR spectra (Fig. 4a). The representative bands of HA and CS denoted by the arrows were evidenced with reference to their pristine materials. The broad peaks around 3367 cm−1 and the weak peak at 3569-cm−1 were assigned to the stretching vibration of OH. The peaks at 2871 and 1080 cm−1 were attributed to the vibration of C–H and the stretching vibration of the C–O in chitosan, respectively. Additionally, the two prominent bands at 1656 cm−1 (amide I band) and 1592 cm−1 (amide II band) were assigned to the C=O stretching vibration of –NHCO– and the N–H bending of –NH2 of CS, respectively. Noticeably, the 1592 cm−1 vibration peak of N–H shifted to 1562 cm−1 in composites, indicating the possible interaction between CS and HA or GO. The peaks at 1413 and 1376 cm−1 originated from the symmetrical deformation modes of CH3 groups in CS, were also detected in the GO–CS–HA composite coatings. Simultaneously, the characteristic split peaks of the PO4 3− cm−1 were observed in HA and composite coatings at 1091 and 1037 cm−1.

FTIR spectrum (a) and Raman spectrum (b) of the GO–CS–HA coatings with different GO contents and pristine GO platelets, HA particles and CS powders
To further confirm the existence of GO and HA in the composite coatings, the Raman analysis was performed (Fig. 4b). As expected, both of the conspicuous and characteristic peaks of GO were observed at 1343 cm−1 (D band, associated with the disorder in GO structure) and 1594 cm−1 (G band, due to the vibration of sp2 carbon atom). The slight increase of the ID/IG ratio from 0.84 in pristine GO to 0.93 in the composite coatings reflected the improvement in the disorder degree. The Raman peaks for HA at 432, 588, 962 and 1045 cm−1 were also detected, which corresponded to the ν2 (PO4 3−), ν4 (PO4 3−), ν1 (PO4 3−) and ν3 (PO4 3−) vibration modes of the phosphate groups. It was no doubt that the GO–CS–HA composite coatings were successfully developed.
Figure 5a demonstrated that the coating mass of CS–HA, 1.0 GO–CS–HA and 1.7 GO–CS–HA were 0.54, 1.58 and 1.70 mg/cm2, respectively. Comparing to CS–HA, the deposition yield was significantly improved by nearly 3 times in 1.0 GO–CS–HA, and even higher by increasing the GO content to 1.7 %. It indicated that more CS–HA nanoparticles were co-deposited onto the composite coatings with the help of GO sheets. The deposition mass w can be estimated by Hamaker’s law [41, 50] as follows:
where C is the concentration of the particle, ε 0 is the permittivity, ε γ is the relative permittivity of the solvent, ξ is the zeta potential of the particles, η is the viscosity of the solvent, E is the applied potential, L is the distance between the electrodes, and t is the deposition time.

The coating mass versus GO content (a) and TG data (b) for the pristine materials CS, HA, GO, and the CS–GO, 1.0 GO–CS–HA and 1.7 GO–CS–HA composite coatings
According to Eq. (1), zeta potential is proportional to the deposition yield. Since GO increased zeta potentials of CS–HA suspensions, the deposition yield was increased too. This finding is in consistence with our previous studies [50], and also in line with Ordikhani et al. [25]. They also reported that the addition of GO increased the deposition rate of CS.
Figure 5b depicted the TG curves of the prepared coatings, and pure HA nanoparticles, GO platelets and CS powders. The weight losses of all samples at low temperature (nearly 100 °C) were ascribed to the evaporation of water content. GO underwent a substantial weight loss at 200 °C related to the decomposition of oxygen-containing functional groups, while CS displayed a gradual weight loss resulted from thermal degradation at a wide temperature range (250–400 °C). In contrast, the HA nanoparticles showed the lowest weight loss due to its high stability. At 500 °C, the residual contents of the CS–HA, 1.0 GO–CS–HA and 1.7 GO–CS–HA composite coatings were 67.3, 80.2 and 82 %, respectively. Given that the main weight loss of the composite coatings originated from the decomposition of CS and GO, these results indicated there were higher amount of HA incorporated in the GO–CS–HA composites than CS–HA composites. Overall, 1.7 GO–CS–HA coating was selected for the following characterization because it displayed uniform morphology and it showed most hydrophilic properties.
3.4 Corrosion property of the coated samples
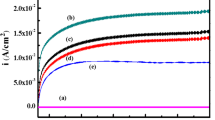
For metallic implant, it is of great importance to improve corrosion resistance to eliminate the metal ion release which could induce adverse biological responses, such as cytotoxicity and metal sensitivity. The corrosion behaviors of the samples were studied by electrochemical potential dynamic polarization tests in SBF as shown in Fig. 6. Corrosion current density (Icorr) of 1.7 GO–CS–HA coating was 13.5 nA/cm2, whereas CS–HA and uncoated Ti exhibited Icorr values of 248 and 6823 nA/cm2, respectively. Therefore, the incorporation of GO sheets into the CS–HA coatings could provide effective corrosion protection for the Ti substrate in simulated physiological environment. This result was in agreement with the HA–CS–MWCNT (multiwalled carbon nanotubes) coating modified on Ti. Rath et al. proposed that the CNT, CS and HA component in the coating could act as barriers to electrons and ions transport between the substrate and SBF, thereby, preventing Ti substrate from corrosion [51]. Specifically, it has been reported that the GO sheets could function as robust corrosion protection barriers to ionic transport in aqueous environment due to its impermeability to small molecules [52]. As an underlying coating, GO improved anti-corrosion performance of organic top-coated steel when a limited amount of the corrosive electrolyte passed through the organic top coating [53]. The corrosion rate exhibited by GO reinforced composite coating on copper was an order of magnitude lower than that of bare copper [54]. This GO–CS–HA composite could be further explored as the protective coating for other metallic substrates with less corrosion resistance, such as stainless steel.

Potentiodynamic polarization plots for the bare Ti substrate and coated samples
3.5 In vitro biocompatibility evaluation
The cell proliferation rate was investigated using CCK-8 assay on the human osteoblast-like MG63 cell line cultured on pure Ti and different EPD surfaces (HA, CS–HA and 1.7 GO–CS–HA) for 1, 3, 5 days. As shown in Fig. 7a, 1.7 GO–CS–HA coating exhibited lowest cell viability around 60 % after cultivation for 1 and 3 days, and then the cell viability increased to 80 % after 5 days of culture. HA coating showed a positive effect on cell proliferation, but the cell viability of CS–HA coating slightly decreased, especially at 1 and 3 days. It was reported that CS coating showed relatively low cell numbers at the early days, but increased cell proliferation after 7 days [55]. Zuo et al. [35] observed a large amount of dead C3H10T1/2 cells on pure CS coating. Meanwhile, there are controversial reports on biocompatibility of graphene-based composites. For example, graphene could alter nanoscale topography and impart more suitable locations for osteoblasts to grow [19]. GO coatings promoted MC3T3-E1 cell differentiation [56]. Depan et al. [57] probed the pre-osteoblast activity on HAp-CS–GO hybrid composites via mineralization of the CS–GO substrate, and found that the synergistic effects of CS and HA (biomineralization) contributed to MC3T3-E1 cellular functions, including spreading, proliferation, differentiation, and mineralization, as well as cell attachment-related protein expression. Also, there were reports showing adverse effect of GO to MG63 cells [58, 59], mammalian fibroblasts [60]. That might be explained by the common toxicity mechanism of carbon-based reactive oxygen species. Especially, the GO physical state, concentration and the cell types have a great impact on their biologic responses. Herein, GO–CS–HA composite coating in this work displayed an acceptable cytotoxicity with low negative effect derived from the CS and GO in the early days.

The cell proliferation rate a and ALP activities b of MG63 cell lines cultured on the pristine Ti and EPD surfaces. SEM images c of pristine Ti (c-1) and EPD surface HA (c-2), CS–HA (c-3), 1.7 GO–CS–HA (c-4) after being immersed in 1.5 SBF for 5 days
Alkaline phosphate (ALP) is an important feature of osteoblast cells expressed in their differentiation phase and a significant quantitative marker of osteogenesis. Figure 7b showed the normalized ALP activities of M63 cells after being cultured on different surfaces for 7 days. The results implied that there was no obvious difference between variable coatings and an elevated ALP expression observed in comparison with pristine Ti. Thus, we concluded that the 1.7 GO–CS–GO composite coating induced no significant inhibitory effect on cell proliferation and differentiation.
The ability of apatite formation on implant surface in SBF could predict in vivo bioactivity of materials [37]. As shown in the SEM images (Fig. 7c-2, c-3, c-4) of EPD coatings, HA, CS–HA and 1.7 GO–CS–HA were completely covered by a flake-like apatite aggregates after being immersed in 1.5 SBF for 5 days, however, there was no obvious deposits on pure Ti surface (Fig. 7c-1). Basically, apatite formation in SBF is achieved by accelerating heterogeneous crystallization in a supersaturated solution towards apatite [61]. It has been assumed that HA surface was immediately negative charged after soaking in SBF and then specifically attracted Ca2+ ions, thereby started the process of apatite formation [62]. Additionally, the dissolution of calcium and phosphate ions from HA led to a higher degree of supersaturation in the local areas and promoted the precipitation of apatite crystal [63]. GO and CS–GO have been widely explored as templates materials for biomineralization. GO could greatly promote nucleation and crystallization of flake-like HA [57, 64]. The negative charged surfaces of GO with deprotonation of –COOH and –OH groups are prone to drive the positive charged Ca2+ ions towards them. Afterwards, PO4 3− ions combined with Ca2+ ions and subjected to the bio-mineralization process. Similarly, the presence of oxygen and amide functional groups in CS could also trigger nucleation of HA through electrostatic interaction. Therefore, CS and GO in CS–HA and 1.7 GO–CS–HA composite coatings further provided nucleating sites for apatite [51, 65]. Overall, the 1.7 GO–CS–HA coating showed the ability to form apatite layer on its surface, possessing the potential to promote bone bonding with the implant.
3.6 Antibacterial adhesion assay
Microbial infection of implants is a severe clinical problem that could lead to implant failure, especially when the biofilm is formed. It is well-known that the HA were favorable for organisms (cell or bacteria) adhesion, due to its good affinity to cell surface proteins and lipids [66]. Since CS and GO could show antibacterial properties, their composite coating was subjected to antibacterial attachment evaluation. This was evaluated by observing the number and morphology of adherent S. aureus bacteria. As shown in Fig. 8a, the largest number of S. aureus cells with round shape morphology were observed on the HA coating. In contrast, the number of adherent bacterial cells decreased remarkably on the CS–HA and 1.7 GO–CS–HA coatings, which was consistent with the result of the bactericidal CS–graphene oxide layers [67]. The speculated antibacterial adhesion mechanism of the nanocomposites was illustrated in Fig. 8b. On the one hand, as natural polysaccharide, chitosan exhibits antibacterial effects against different groups of microorganisms, including fungi and bacteria. It has been proposed that the interaction between positively charged chitosan molecules and negatively charged microbial cell membrane may result in leakage of proteinaceous and other intracellular constituents [68]. On the other hand, graphene-based materials also exhibit bactericidal property due to their ability to disrupt and damage cell membranes intact through membrane and oxidative stress [69, 70]. Besides, it has been reported that S. aureus as a Gram-positive bacteria is more sensitive to the direct contact interaction with graphene-oxide sheets than Gram-negative bacteria [71].

SEM images a of the S. aureus after incubation 12 h with the pristine Ti (1) and different coating interfaces: HA (2), CS–HA (3), 1.7 GO–CS–HA (4). The black arrows in (3), (4) point to the adherent S. aureus. Scale bar 3 μm. Schematic illustration b of the speculated antibacterial adhesion mechanism of the GO–CS–HA nanocomposites
For future researches to further improve coating anti-bacteria properties, it will be feasible to utilize CS to absorb antibacterial metal ions, such as silver, copper or zinc [52]. Simultaneously, GO nanosheets could be explored as barriers to control the release of these antibacterial molecules or ions.
4 Conclusions
GO–CS–HA nanocomposites were successfully deposited on Ti substrate through EPD method. The addition of GO into CS–HA increased the suspension stability and promoted HA deposition. The resulting GO–CS–HA coatings could improve hydrophilicity of the surfaces and provide effective corrosion protection of the Ti substrate. After 5 days incubation, the ternary composite coating induced no significant in vitro cytotoxicity towards MG63 cells. And there was significant apatite formation on the coating in 1.5 SBF. More importantly, the GO–CS–HA nanocomposite coating could reduce the adhesion of bacteria onto titanium. Thus this nanocomposite coating fabricated by EPD showed promising potential on surface functionalization of bone-related orthopedic and dental implants.
References
Boccaccini AR, Keim S, Ma R, et al. Electrophoretic deposition of biomaterials. J R Soc Interface. 2010;7:S581–613.
Fukada Y, Nicholson PS, et al. Electrophoretic deposition-mechanisms, myths and materials. J Mater Sci. 2004;39:787–801.
Woodard JR, Hilldore AJ, Lan SK, et al. The mechanical properties and osteoconductivity of hydroxyapatite bone scaffolds with multi-scale porosity. Biomaterials. 2007;28:45–54.
Ma R, Li Y, Zhitomirsky I. Electrophoretic deposition of hyaluronic acid and composite films for biomedical applications. JOM. 2010;62:72–5.
Pang X, Zhitomirsky I. Electrodeposition of composite hydroxyapatite–chitosan films. Mater Charact. 2007;58:339–48.
Ma R, Epand RF, Zhitomirsky I. Electrodeposition of hyaluronic acid and hyaluronic acid–bovine serum albumin films from aqueous solutions. Colloid Surf B. 2010;77:279–85.
Cheong M, Zhitomirsky I. Electrodeposition of alginic acid and composite films. Colloid Surf A. 2008;328:73–8.
Sun F, Pang X, Zhitomirsky I. Electrophoretic deposition of composite hydroxyapatite–chitosan–heparin coatings. J Mater Process Technol. 2009;209:1597–606.
Lahiji A, Sohrabi A, Hungerford DS, et al. Chitosan supports the expression of extracellular matrix proteins in human osteoblasts and chondrocytes. J Biomed Mater Res. 2000;51:586–95.
Frohbergh ME, Katsman A, Botta GP, et al. Electrospun hydroxyapatite-containing chitosan nanofibers crosslinked with genipin for bone tissue engineering. Biomaterials. 2012;33:9167–78.
Chesnutt BM, Yuan Y, Buddington K, et al. Composite chitosan/nano-hydroxyapatite scaffolds induce osteocalcin production by osteoblasts in vitro and support bone formation in vivo. Tissue Eng A. 2009;15:2571–9.
Wang Y, Li Z, Wang J, et al. Graphene and graphene oxide: biofunctionalization and applications in biotechnology. Trends Biotechnol. 2011;29:205–12.
Eissa S, Jimenez GC, Mahvash F, et al. Functionalized CVD monolayer graphene for label-free impedimetric biosensing. Nano Res. 2014;8:1698–709.
Liu J, Cui L, Losic D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013;9:9243–57.
Imani R, Emami SH, Faghihi S. Synthesis and characterization of an octaarginine functionalized graphene oxide nano-carrier for gene delivery applications. Phys Chem Chem Phys. 2015;17:6328–39.
Song F, Hu W, Xiao L, et al. Enzymatically cross-linked hyaluronic acid/graphene oxide nanocomposite hydrogel with pH-responsive release. J Biomater Sci Polym E. 2015;26:339–52.
Liao JF, Qu Y, Chu BY, et al. Biodegradable CSMA/PECA/graphene porous hybrid scaffold for cartilage tissue engineering. Sci Rep. 2015;5:1–16.
Srivastava S, Kumar V, Ali MA, et al. Electrophoretically deposited reduced graphene oxide platform for food toxin detection. Nanoscale. 2013;5:3043–51.
Zhang L, Liu W, Yue C, et al. A tough graphene nanosheet/hydroxyapatite composite with improved in vitro biocompatibility. Carbon. 2013;61:105–15.
Fan Z, Wang J, Wang Z, et al. One-pot synthesis of graphene/hydroxyapatite nanorod composite for tissue engineering. Carbon. 2014;66:407–16.
Baradaran S, Moghaddam E, Basirun WJ, et al. Mechanical properties and biomedical applications of a nanotube hydroxyapatite-reduced graphene oxide composite. Carbon. 2014;69:32–45.
Neelgund GM, Oki A, Luo Z. In situ deposition of hydroxyapatite on graphene nanosheets. Mater Res Bull. 2013;48:175–9.
Janković A, Eraković S, Mitrić M, et al. Bioactive hydroxyapatite/graphene composite coating and its corrosion stability in simulated body fluid. J Alloy Compd. 2015;624:148–57.
Nair M, Nancy D, Krishnan AG, et al. Graphene oxide nanoflakes incorporated gelatin–hydroxyapatite scaffolds enhance osteogenic differentiation of human mesenchymal stem cells. Nanotechnology. 2015;26:161001–10.
Ordikhani F, Farani MR, Dehghani M, et al. Physicochemical and biological properties of electrodeposited graphene oxide/chitosan films with drug-eluting capacity. Carbon. 2015;84:91–102.
Fan H, Wang L, Zhao K, et al. Fabrication, mechanical properties, and biocompatibility of graphene-reinforced chitosan composites. Biomacromolecules. 2010;11:2345–51.
Li M, Wand YB, Cheng Y. In situ synthesis and biocompatibility of nano hydroxyapatite on pristine and chitosan functionalized graphene oxide. J Mater Chem B. 2013;1:475–84.
Liu H, Cheng J, Chen F, et al. Gelatin functionalized graphene oxide for mineralization of hydroxyapatite: biomimetic and in vitro evaluation. Nanoscale. 2014;6:5315–22.
Deepachitra R, Nigam R, Purohit SD, et al. In vitro study of hydroxyapatite coatings on fibrin functionalized/pristine graphene oxide for bone grafting. Mater Manuf Process. 2015;30:804–11.
de Moraes ACM, Andrade PF, de Faria AF, et al. Fabrication of transparent and ultraviolet shielding composite films based on graphene oxide and cellulose acetate. Carbohydr Polym. 2015;123:217–27.
Shin YC, Lee JH, Jin L, et al. Stimulated myoblast differentiation on graphene oxide-impregnated PLGA-collagen hybrid fiber matrices. J Nanobiotechnol. 2015;13:21–31.
Rodríguez-Lozano FJ, García-Bernal D, Aznar-Cervantes S, et al. Effects of composite films of silk fibroin and graphene oxide on the proliferation, cell viability and mesenchymal phenotype of periodontal ligament stem cells. J Mater Sci Mater Med. 2014;25:2731–41.
He Y, Zhang N, Gong Q, et al. Alginate/graphene oxide fibers with enhanced mechanical strength prepared by wet spinning. Carbohydr Polym. 2012;88:1100–8.
Yang X, Tu Y, Li L, et al. Well-dispersed chitosan/graphene oxide nanocomposites. Acs Appl Mater Interfaces. 2010;2:1707–13.
Zuo PP, Feng HF, Xu ZZ, et al. Fabrication of biocompatible and mechanically reinforced graphene oxide-chitosan nanocomposite films. Chem Cent J. 2013;7:39–49.
Liu L, Li C, Bao C, et al. Preparation and characterization of chitosan/graphene oxide composites for the adsorption of Au(III) and Pd (II). Talanta. 2012;93:350–7.
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27:2907–15.
Mohammadi MR, Ordikhani F, Fray DJ, et al. Template-based growth of titanium dioxide nanorods by a particulate sol-electrophoretic deposition process. Particuology. 2011;9:161–9.
Fang M, Long J, Zhao W, et al. pH-responsive chitosan-mediated graphene dispersions. Langmuir. 2010;26:16771–4.
Cote LJ, Kim J, Tung VC, et al. Graphene oxide as surfactant sheets. Pure Appl Chem. 2010;83:95–110.
Besra L, Liu M. A review on fundamentals and applications of electrophoretic deposition (EPD). Prog Mater Sci. 2007;52:1–61.
Pang X, Zhitomirsky I. Electrodeposition of composite hydroxyapatite–chitosan films. Mater Chem Phys. 2005;94:245–51.
Moon IK, Kim JI, Lee H, et al. 2D graphene oxide nanosheets as an adhesive over-coating layer for flexible transparent conductive electrodes. Sci Rep. 2013;3:1–7.
Ganesh BM, Isloor AM, Ismail AF. Enhanced hydrophilicity and salt rejection study of graphene oxide-polysulfone mixed matrix membrane. Desalination. 2013;313:199–207.
Gonçalves G, Marques PAAP, Barros-Timmons A, et al. Graphene oxide modified with PMMA via ATRP as a reinforcement filler. J Mater Chem. 2010;20:9927–34.
Nosonovsky M, Bhushan B. Roughness optimization for biomimetic superhydrophobic surfaces. Microsyst Technol. 2005;11:535–49.
Zuldesmi M, Waki A, Kuroda K, et al. Hydrothermal treatment of titanium alloys for the enhancement of osteoconductivity. Mat Sci Eng C. 2015;49:430–5.
Arima Y, Iwata H. Effect of wettability and surface functional groups on protein adsorption and cell adhesion using well-defined mixed self-assembled monolayers. Biomaterials. 2007;28:3074–82.
Gittens RA, Scheideler L, Rupp F, et al. A review on the wettability of dental implant surfaces II: biological and clinical aspects. Acta Biomater. 2014;10:2907–18.
Li M, Liu Q, Cheng Y, et al. Electrophoretic deposition and electrochemical behavior of novel graphene oxide-hyaluronic acid-hydroxyapatite nanocomposite coatings. Appl Surf Sci. 2013;284:804–10.
Rath PC, Singh BP, Besra L, et al. Multiwalled carbon nanotubes reinforced hydroxyapatite-chitosan composite coating on Ti metal: corrosion and mechanical properties. J Am Ceram Soc. 2012;95:2725–31.
Kirkland NT, Schiller T, Medhekar N, et al. Exploring graphene as a corrosion protection barrier. Corros Sci. 2012;56:1–4.
Park JH, Park JM. Electrophoretic deposition of graphene oxide on mild carbon steel for anti-corrosion application. Surf Coat Technol. 2014;254:167–74.
Singh BP, Nayak S, Nanda KK, et al. The production of a corrosion resistant graphene reinforced composite coating on copper by electrophoretic deposition. Carbon. 2013;61:47–56.
Pishbin F, Mouriño V, Gilchrist JB, et al. Single-step electrochemical deposition of antimicrobial orthopaedic coatings based on a bioactive glass/chitosan/nano-silver composite system. Acta Biomater. 2013;9:7469–79.
Zhao C, Lu X, Zanden C, et al. The promising application of graphene oxide as coating materials in orthopedic implants: preparation, characterization and cell behavior. Biomed Mater. 2015;10:1–9.
Depan D, Pesacreta TC, Misra RDK. The synergistic effect of a hybrid graphene oxide–chitosan system and biomimetic mineralization on osteoblast functions. Biomater Sci. 2014;2:264–74.
Li M, Liu Q, Cheng Y, et al. Graphene oxide/hydroxyapatite composite coatings fabricated by electrophoretic nanotechnology for biological applications. Carbon. 2014;67:185–97.
Lahiri D, Dua R, Zhang C, et al. Graphene nanoplatelet-induced strengthening of ultrahigh molecular weight polyethylene and biocompatibility in vitro. Acs Appl Mater Interfaces. 2012;4:2234–41.
Liao KH, Lin YS, Macosko CW, et al. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. Acs Appl Mater Interfaces. 2011;3:2607–15.
Bohner M, Lemaitre J. Can bioactivity be tested in vitro with SBF solution? Biomaterials. 2009;30:2175–9.
Kim HM, Himeno T, kawashita M, et al. The mechanism of bio mineralization of bone-like apatite on synthetic hydroxyapatite: an in vitro assessment. J R Soc Interface. 2004;1:17–22.
Juhasz JA, Best SM, Auffret AD, et al. Biological control of apatite growth in simulated body fluid and human blood serum. J Mater Sci Mater Med. 2008;19:1823–9.
Gao F, Xu C, Hu H, et al. Biomimetic synthesis and characterization of hydroxyapatite/graphene oxide hybrid coating on Mg alloy with enhanced corrosion resistance. Mater Lett. 2015;138:25–8.
Núñez JD, Benito AM, González R, et al. Integration and bioactivity of hydroxyapatite grown on carbon nanotubes and graphene oxide. Carbon. 2014;79:590–604.
Liu Y, Huang J, Ding S, et al. Deposition, characterization, and enhanced adherence of Escherichia coli bacteria on flame-sprayed photocatalytic titania-hydroxyapatite coatings. J Therm Spray Technol. 2013;22:1053–62.
Mazaheri M, Akhavan O, Simchi A. Flexible bactericidal graphene oxide–chitosan layers for stem cell proliferation. Appl Surf Sci. 2014;301:456–62.
Rabea EI, Badawy MET, Stevens CV, et al. Chitosan as antimicrobial agent: applications and mode of action. Biomacromolecules. 2003;4:1457–65.
Liu S, Zeng TH, Hofmann M, et al. Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: membrane and oxidative stress. ACS Nano. 2011;5:6971–80.
Chen J, Peng H, Wang X, et al. Graphene oxide exhibits broad-spectrum antimicrobial activity against bacterial phytopathogens and fungal conidia by intertwining and membrane perturbation. Nanoscale. 2014;6:1879–89.
Akhavan O, Ghaderi E. Toxicity of graphene and graphene oxide nanowalls against bacteria. ACS Nano. 2010;4:5731–6.
Acknowledgments
This work is jointly supported by the Project of Scientific and Technical Plan of Beijing (No. Z141100002814008), National Natural Science Foundation of China (No. 31370954), State Key Laboratory of Bioelectronics Open Research Fund of China (Chien-Shiung Wu Laboratory), National Basic Research Program (973) of China (2012CB619102).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shi, Y.Y., Li, M., Liu, Q. et al. Electrophoretic deposition of graphene oxide reinforced chitosan–hydroxyapatite nanocomposite coatings on Ti substrate. J Mater Sci: Mater Med 27, 48 (2016). https://doi.org/10.1007/s10856-015-5634-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-015-5634-9