
Abstract
The paper reports on a simple method of synthesizing [PEO(PCL)2] triarm star-shaped copolymers by a combination of Michael-addition type reaction and ring-opening polymerization. A Michael-addition reaction yielded a PEO end-capped by two hydroxyl groups—a [PEO(OH)2] macroinitiator—which was used for sequential building of PCL blocks. The macroinitiator and copolymers were analyzed by FTIR, 1H NMR spectroscopy and SEC. The self-assembly behavior of the copolymers in aqueous media was studied by UV–Vis spectroscopy. The size and morphology of the obtained micelles were determined by TEM. None of the polymers had cytotoxic effects in vitro. Cellular uptake studies showed the accumulation of neutral red loaded micelles in the perinuclear area of human hepatocellular carcinoma cells revealing a cellular uptake associated with macropinocytosis and caveolae mediated endocytosis. The accumulation had a sustained effect over 3 days pointing at the potential application of the copolymers micelles as a drug delivery system.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Recently block copolymers of complex architecture have been receiving a great attention [1–8]. Amongst them star-shaped block copolymers consisting of at least three linear polymeric chains (arms) with radial arrangement around a central molecular fragment (core) [9–11] appear to be the most promising for various emerging applications owing to their smaller hydrodynamic volume and radius of gyration, and lower melt and solution viscosities compared with their linear counterparts [3, 12–16].
Star-shaped polymers are usually prepared by the “arm-first” or “core-first” method. The “arm-first” route involves construction of polymer arms on a macroinitiator that contains a precise number of reactive sites [17, 18]. The “core-first” approach utilizes multifunctional low molecular weight initiators allowing the synthesis of block copolymer chains [19]. Several synthetic strategies have been described for the preparation of star-shaped block copolymers based on poly(ethylene oxide) (PEO) and polyesters such as poly(ε-caprolactone) (PCL) [4, 8, 20] or polylactides (PLA) [21–23]. It has been found that block copolymers composed of PCL and PEO introduced into the body are degraded into non-toxic and readily excreted products [24, 25]. Indeed, PEO is an excellent biocompatible material owing to its flexibility, non-toxicity and hydrophilicity [26]. PCL is a non-toxic biodegradable polymer of a hydrophobic character [27]. The different solubility of PEO and PCL blocks determines the specific and unique solution behavior of these copolymers [8]. As it is well-known, block copolymers of this kind are able to form nanosized micelles with a core–shell architecture. These envelopes have great potential as drug delivery systems because their hydrophobic cores can be used as reservoirs for lipophilic agents (drugs). Being able to protect drugs from degradation as well as to reduce drugs toxicity polymeric micelles composed of PEO–PCL block copolymers have previously been used as drug carriers. They also could minimize or even remove some of the unwanted side effects [28–33]. A wide variety of in vitro assays has been successfully used to screen the potential role of polymeric micelles as drug carriers. Many different cell lines are commonly used as model systems when studying the properties of such nanoparticles. In most cases the function of the selected cells is detrimentally affected by their uptake. The phenomenon is observed especially with hepatocytes which are the most abundant cell type within the liver and are responsible for the majority of liver functions [34].
In this paper we present a new, simple and efficient route to synthesis of AB2 triarm amphiphilic block copolymers, consisting of hydrophilic PEO (A) and hydrophobic PCL (B) arms. The advantage of this approach is both high yields and excellent purity of the obtained polymer products. In this study we also demonstrate the ability of the star-shaped copolymers to form nanosized micelles as well as their capability to deliver a low molecular organic substance into living cells.
2 Experimental
2.1 Materials
ε-Caprolactone (ε-CL, 99%, CAPA®) was kindly provided by Solvay (UK). The ε-CL was dried over calcium hydride with continuous stirring at room temperature for 48 h and distilled under reduced pressure before use. Poly(ethylene oxide) methyl ether methacrylate (MePEOMA, 1,100 g/mol), 1,6-diphenyl-1,3,5-hexatriene (DPH, 98%), Neutral Red (NR), Tin (II) 2-ethylhexanoate (Sn(Oct)2, 95%, 0.06 M solution in toluene), and phosphate buffered saline (PBS, P5368) were received from Sigma-Aldrich. 3-mercapto-1,2-propanediol (~98%, Fluka), DPH, and NR were used without further purification. Pyridine (~99%, Aldrich) was dried over calcium hydride and distilled under reduced pressure. Toluene (99%, Labscan) was refluxed for 24 h over Na–K alloy under dry argon atmosphere and then distilled. Tetrahydrofuran (THF, 99%, Fluka) was dried by refluxing over calcium hydride for 24 h and distilled under argon atmosphere. All aqueous solutions were prepared in doubly distilled water (ddH2O).
2.2 Cell line, reagents and chemicals
Human liver hepatocellular carcinoma cell line (HepG2) was obtained from American Type Culture Collection (HB-8065) and maintained in DMEM (Dilbecco’s Modified Eagle Medium, Applichem, Germany) supplemented with 10% (v/v) FBS (Fetal Bovine Serum, BioWhittaker, USA), penicillin (100 μg/ml), streptomycin (100 μg/ml) and 4.0 mM l-glutamine at 37 °C in a humidified atmosphere of 5.0% CO2 and 95% air. Cells were routinely checked for mycoplasma contamination, by 4′,6-diamidino-2-phenylindole staining (DAPI, Roche Diagnostics, Germany) and found free of it. For subsequent experiments, HepG2 cells were grown in tissue culture dishes or multiwell plates (Nunc, Germany).
2.3 Synthesis of α-methoxy-ω,ω′-dihydroxy poly(ethylene oxide) macroinitiator [PEO(OH)2], ((2) Scheme 1)
The reaction was carried out as follows: MePEOMA (6.0 g, 5.4 mmol) was placed in a 100 ml one-neck round-bottom flask, dissolved in 30 ml of anhydrous toluene and dried by azeotropic distillation. 3-mercapto-1,2-propanediol (1.1 equiv., 6.0 mmol), pyridine (2.0 equiv.) and dry THF (15 ml) were then added. The reaction mixture was stirred at 25 °C for 3 days under argon atmosphere in the dark, followed by removing of pyridine and THF under reduced pressure. The polymer was dissolved in toluene and precipitated in cooled diethyl ether and then recovered by filtration. The precipitate was washed with diethyl ether and dried in vacuo (Yield: 90%).

Synthetic pathway for the preparation of [PEO(PCL)2] star-shaped block copolymers
2.4 Synthesis of [PEO(PCL)2] star-shaped copolymers, ((3) Scheme 1)
Typically, 0.5 g (1.0 mmol) of [PEO(OH)2] were introduced into a 50 ml glass reactor containing a magnetic bar. The polymer was dissolved in dry toluene and dried three times by azeotropic distillations. A certain amount of freshly distilled ε-CL was then added. After heating, a 0.1 ml of 0.06 M Sn(Oct)2 was rapidly injected through a septum and the polymerization was carried out for 48 h at 110 °C. The reactor was cooled to room temperature and the reaction mixture was dissolved in toluene. The copolymer was collected by precipitation in cooled diethyl ether, filtrated, and dried overnight at 40 °C in vacuum.
2.5 Characterization techniques
All proton nuclear magnetic resonance (1H NMR) spectra were recorded at 250 MHz on a Bruker Avance DRX 250 spectrometer using deuterated chloroform as a solvent and tetramethylsilane as an internal reference.
The Fourier transform infrared (FTIR) spectra were recorded on a Bruker-Vector 22 spectrometer at a resolution of 1–2 cm−1 accumulating 50 scans. The samples were prepared in the form of KBr pellets.
Ultraviolet–visible (UV–Vis) investigations were performed with a Beckman L-80 UV–Vis spectrometer (Beckman-Coulter, USA).
The size and morphology of polymeric micelles were studied by transmission electron microscopy (TEM). The sample of micellar solutions were dropped on formvar-coated copper grids, air dried for several hours at room temperature followed by drying in a vacuum oven for 2 h at 40°C. The experiments were performed on a JEOL (JEM-100B) microscope with accelerating voltage of 80 kV.
The number average molecular weight (M n ), weight average molecular weight (M w ), and polydispersity index (M w /M n ) of prepared macroinitiator and block copolymers were determined on a size exclusion chromatography (SEC) system (Waters) equipped with a double detection—differential refractometer M410 and a M484 UV detector. Data collection and processing were done using a Clarity software. The analyses were performed on Ultrastyragel Linear, Styragel 100 Å, and Styragel 500 Å columns (Waters) calibrated with PEO standards. THF was used as a mobile phase at a flow rate of 1.0 ml/min at 45 °C.
2.6 Formation of polymeric micelles and determination of the critical micelle concentration of [PEO(PCL)2] block copolymers
The micelles of [PEO(PCL)2] block copolymers (3a, 3b, and 3c, in Table 2) were prepared by the following procedure. A sample of [PEO(PCL)2] triarm block copolymer (5.0 mg) was initially dissolved in 1.0 ml THF and then made up to 100 ml with ddH2O under vigorous stirring at 5 °C. After that, the organic co-solvent was removed by a rotary evaporator at 30 °C for 1 h and a series of [PEO(PCL)2] aqueous solutions of different concentration (0.05 to 1.0 × 10−5 g/L) were prepared. The critical micelle concentration (CMC) of the [PEO(PCL)2] triarm block copolymers was determined using DPH as a hydrophobic probe. The obtained series of copolymer solutions were added to vials containing methanol solution of DPH. The final concentration of DPH was 4.0 × 10−6 M. All samples were kept in the dark at room temperature for 24 h to reach the solubilization equilibrium of DPH in the aqueous phase before further analysis.
2.7 Formation of NR-loaded [PEO(PCL)2] block copolymer micelles
The loading of NR into the polymeric micelles was realized by diluting a stock solution of NR (6.5 mM) with a predetermined volume of 10.0 μM aqueous [PEO(PCL)2] solution. The final concentration of NR in the micellar solution was 0.376 mM. The resulting solution was shaking overnight at room temperature and allowed to stand stationary for additional 24 h.
2.8 Cytotoxicity assay
The in vitro cytotoxicity assay (methyl-thiazol-tetrazolium based, MTT assay) was performed according to Mosmann [35]. Briefly, logarithmic growing HepG2 cells were harvested and plated at a density of 5.0 × 104 cells/well in 100 μl DMEM. Twenty-four hours later the different concentrations (4.3; 8.6; 21.5 μM) of micellar solutions were added and incubated for 24, 48, and 72 h. Following the incubation periods 10 μl of MTT solution (5.0 mg/ml) was added and re-incubated for further 3 h. The MTT-formazan product was dissolved in isopropanol and the absorption at 550/630 nm was measured on an ELISA plate reader (Bio-Tek Instruments Inc., USA). MTT assay has been performed eight times in total.
2.9 Cellular uptake of NR-loaded polymeric micelles
HepG2 cells were seeded into 24-well plates at a density of 5.0 × 104 cells/well in 200 μl DMEM media. After 24 h incubation, DMEM media was removed and fresh solutions of NR and NR-loaded micelles were added. For this purpose, the NR solution in ddH2O and NR-loaded micelles were diluted with equivalent volumes of cell culture medium. The NR concentration in all wells was 0.138 mM, whereas the polymer concentration was 5.0 μM. Cells were further incubated for 4, 24, 48, and 72 h. At the end of the incubation time cells were washed three times with PBS and fixed with 1.0% formaldehyde. The NR intensity inside cells was evaluated after extraction with 1.0% glacial acetic acid in 50% ethanol. The NR accumulated in HepG2 cells was quantified spectrophotometrically at 540 nm with DTX880 (Becman-Coulter, USA) against cells incubated only with NR.
For light microscopy studies, the cells were seeded on a sterile round glass cover slips at same conditions mentioned above. At the end of incubation period, the medium was withdrawn, the cells were washed with PBS and fixed with a 4.0% of paraformaldehyde in PBS. Cover slips were mounted on glass slides with UltraCruz mounting media (SantaCruz, USA) and observed under Telaval microscope (Carl Zeiss, Germany).
2.10 In vitro NR-release study
The release of NR from the NR-loaded micelles has been studied in vitro following 24 h incubation of HepG2 cells with the same concentration as in the celllular uptake study. Following this period the cells were washed three times with PBS and incubated up to 200 h with fresh DMEM. Quantification of the released NR was performed specrophotometrically and was normalized against the cell numbers obtained by direct counting with an automated cell counter (Countess, Invitrogen, USA).
2.11 Endocytosis pathway analysis
The HepG2 cells were incubated for 30 min with different inhibitors—cytochalasin D (CytD, 3.0 μg/ml), chlorpromazine (CPZ, 6.0 μg/ml), cytochalasin B (CytB, 6.0 μg/ml), and methyl-β-cyclodextrin (MβC, 100 μg/ml). Following incubation, the cells have been treated with NR-loaded micelles for 24 h. Subsequently, the cells were washed three times with PBS and fixed with 1.0% formaldehyde. The NR accumulated in the HepG2 cells was quantified spectrophotometrically at 540 nm with DTX880.
2.12 Statistical analysis
Data are reported as mean ± standard deviation (SD). Differences between the means were determined by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons using PASW Statistic 18 (IBM, Chicago, USA). The level of significance was set at p = 0.05.
3 Results and discussion
3.1 Synthesis of [PEO(OH)2] macroinitiator (compound 2, Scheme 1)
The Michael-addition type reaction was used as a convenient method for the synthesis of α-methoxy-ω,ω′-dihydroxy poly(ethylene oxide) [PEO(OH)2]. The [PEO(OH)2] was designed as a macroinitiator for the sequential ring-opening polymerization (ROP) of ε-CL (Scheme 1). For this purpose, commercially available MePEOMA (electron withdrawing enone) with molecular weight 1,100 g/mol, was reacted with 3-mercapto-1,2-propanediol (nucleophile). This type of reaction typically refers to the base-catalyzed addition of nucleophiles, such as thiols, to an activated α,β-unsaturated carbonyl-containing compound. The reaction was performed under mild reaction conditions (room temperature, dry argon atmosphere, polar aprotonic solvent THF, and in the dark) in the presence of pyridine, as a highly active homogeneous base catalyst. The macromonomer (MePEOMA) conversion estimated from the relative intensity of the signals characteristic of both H2C=C– (δ = 5.57 ppm) and CH3O– (δ = 3.39 ppm) groups in the 1H NMR spectrum was found to be quantitative (~85% at reaction time 72 h). After purification, the structure of the macroinitiator was confirmed by 1H NMR and FTIR spectroscopy. [PEO(OH)2] M n was determined by 1H NMR spectroscopy and SEC. The macromolecular characteristics of MePEOMA and [PEO(OH)2] macroinitiator are listed in Table 1.
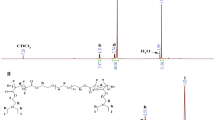
The structural features of the synthesized [PEO(OH)2] macroinitiator were determined by 1H NMR spectroscopy (Fig. 1). The broad signal marked b at 3.65 ppm is characteristic of the methylene protons of ethylene oxide (EO) repeating units. Other signals in the vicinity of the one at 3.65 ppm have been also observed, namely: at 2.76 ppm typical of –(CH3)CH– (e); at 2.74 and 2.51 ppm characteristic of –S–CH2–CH(OH)– (f) and at 2.69 and 2.93 ppm ascribed to –CH(CH3)–CH2–S–(g). The signal observed at 1.61 ppm (d) is assigned to the three protons of –CH3 group, and a singlet signal detected at 3.39 ppm (peak a)—for the α-methoxy protons. Figure 1 also shows a complex signal, observed at high chemical shifts which corresponds to the two protons from –CH 2–O–C(O) at 4.25 ppm (peak c). Unfortunately, the spectrum of [PEO(OH)2] macroinitiator did not permit a clear determination of the signals labeled h and i (Fig. 1) because they are hidden in the broad signal of the EO repeating units at 3.65 ppm. [PEO(OH)2] M n (2, in Table 1) was determined by 1H NMR spectroscopy and calculated according to Eq. 1:
where I b and I a are the integral values of the signals at 3.65 ppm (PEO) and 3.39 ppm (methoxy end-group), M RU is the molecular weight of the repeating unit (–CH2–CH2–O–), while M EG1 and M EG2 stand for the molecular weights of the (–S–CH2–CH(OH)–CH2–OH) and (CH3O–) end groups.

1H NMR spectrum of [PEO(OH)2] (2, in Table 1)
The SEC chromatograms of starting MePEOMA and of the corresponding [PEO(OH)2] macroinitiator show a monomodal distribution (Fig. 1 in Supplementary Data). Naturally, the elution time of [PEO(OH)2] macroinitiator was shifted towards lower values corresponding to a higher molecular weight if compared to those of the starting macromonomer. Besides, the analysis clearly demonstrates the absence of any side products of lower or higher M n values formed as a result of the telomerization of MePEOMA, of the hydrolysis or transesterification of MePEOMA ester bonds.
The chemical composition and structure of the macroinitiator were further characterized by FTIR spectroscopy (Fig. 2a). The presence of hydroxyl groups at ω and ω′ positions in the structure of [PEO(OH)2] was proven by the appearance of a broad and intense absorption band with a maximum at 3,501 cm−1 (Fig. 2a). An analogous band was also observed in the spectrum of 3-mercapto-1,2-propanediol (Fig. 2c) at 3,375 cm−1 indicating the existence of intramolecular hydrogen bonding between the hydroxyl groups and the neighbouring thiol one. The presence of a C–S bond in the macroinitiator structure was evidenced by the appearance of a new low-intensity band at 708 cm−1 assigned to C–S–C stretching vibration, characteristic of thioether compounds. In the FTIR spectrum of [PEO(OH)2] the distinctive bands of aliphatic esters (at 1,731 cm−1), ether groups of the EO repeated units (at 1,105 cm−1), and of primary hydroxyl group (at 1,039 cm−1) were also observed (Fig. 2a). Moreover, the effectiveness of the synthetic procedure (a reaction between the used thiol and MePEOMA) was confirmed by the complete disappearance of the peaks corresponding to the methacrylate H2C=C(CH3)– group at 820 cm−1 (see Fig. 2b) and –SH group at 2,560 cm−1 (see Fig. 2c) in the macroinitiator’s spectrum.

FTIR spectra of the (a) [PEO(OH)2], (b) MePEOMA, and (c) 3-mercapto-1,2-propanediol
3.2 Synthesis of [PEO(PCL)2] star-shaped copolymers (compound 3, Scheme 1)
The [PEO(PCL)2] triarm star-shaped copolymers were successfully synthesized by ROP of the ε-CL monomer. The synthesized [PEO(OH)2] was used as an efficient macroinitiator in the presence of Sn(Oct)2 as a catalyst. The involvement of both terminal and internal hydroxyl groups (primary and secondary alcohol groups) as initiators for ROP of ε-CL monomers has been demonstrated in numerous experimental examples [36, 37]. The length of the PCL blocks was controlled by regulating the ε-CL/[PEO(OH)2] molar ratio. After purification, the [PEO(PCL)2] block copolymers were characterized by 1H NMR, FTIR spectroscopy and SEC analysis.
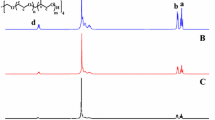
The 1H NMR spectrum of the [PEO(PCL)2] triarm block copolymer (Fig. 3) has the signals characteristic of PCL and PEO chains. The methylene protons of EO repeating units marked with b were observed at 3.66 ppm. The signals of PCL protons were detected at 4.07 ppm (l) (–CH2–OC(O)), 2.32 ppm (h) (–C(O)–CH2), 1.66 ppm (i + j) (–C(O)–CH2–CH2–CH2–CH2–), 1.62 ppm (d) (–CH3), and 1.39 ppm (k) (–C(O)–CH2–CH2–CH2–), respectively. The M n of the [PEO(PCL)2] block copolymers was determined by 1H NMR using the following equation:
where I l and I b stand for the integral values of the methylene protons of the PCL at 4.07 ppm (peak l, Fig. 3) and for the methylene protons of the PEO at 3.66 ppm (peak b, Fig. 3). The molecular weight of a ε-CL monomer unit is 114.14. DPPEO is the degree of polymerization of the PEO and M n(NMR) [PEO(OH)2] is the molecular weight of the macroinitiator. The observed experimental degrees of ε-CL polymerization are in good agreement with the theoretically expected values (Table 2).

1H NMR spectrum for the [PEO(PCL)2] star-shaped copolymer (3b, in Table 2)
The SEC curves of the triarm star-shaped copolymers are monomodal, symmetric and of a narrow molecular weight distribution (Fig. 2 in Supplementary Data). The values for M n and M w /M n for all [PEO(PCL)2] block copolymers are listed in Table 2.
The structure of the obtained [PEO(PCL)2] triarm star-shaped block copolymers was also confirmed by FTIR spectroscopy. The FTIR spectra of all samples show similar bands and signals. The observed characteristic peaks are the following (Fig. 4): at 1,725 cm−1—ester bond (C=O stretching) of the ε-CL repeated units of the PCL blocks; at 1,189 cm−1—ether bond (C–O–C stretching) of the EO repeated units of the PEO backbone; at 2,950 and 2,866 cm−1 –C–H vibrations typical for both monomer units; and at 3,649 cm−1—the characteristic band assigned to the terminal hydroxyl groups in block copolymers.

FTIR spectrum of the [PEO(PCL)2] star-shaped copolymer (3b, in Table 2)
3.3 Micellization of the [PEO(PCL)2] triarm star-shaped block copolymers
We investigated CMC, micelle size, and morphology of the prepared [PEO(PCL)2] star-shaped block copolymers in aqueous media. As known the different water-solubility of PEO and PCL blocks is the factor determining the amphiphilic behavior of the obtained star-shaped block copolymers. Such block copolymers self-assemble in aqueous medium forming nanospheric micelles having a PCL hydrophobic core and a shell of hydrophilic PEO chains [38, 39]. The micellization of [PEO25(PCL8)2], [PEO25(PCL17)2], and [PEO25(PCL26)2] copolymers were monitored by a UV–Vis spectroscopy using DPH as a hydrophobic probe. CMC was determined as a result of the spectral changes in DPH molecules in the UV–Vis region [40]. The data revealed that at low concentrations of the star-shaped block copolymers the DPH absorbance was practically unchanged and close to zero. At a higher concentration of the copolymers the absorbance of DPH changed dramatically in response to alterations in its environmental polarity (entrapping of lipophilic DPH in the hydrophobic micelles cores). CMC values were determined from the intersection of horizontal and vertical tangents of the obtained curves (Fig. 5a). CMC values for [PEO25(PCL8)2], [PEO25(PCL17)2] and [PEO25(PCL26)2] copolymers were found to be 2.80 × 10−3 g/L, 2.14 × 10−3 g/L, and 1.05 × 10−3 g/L, respectively. The CMC value for the [PEO25(PCL35)2] copolymer was not determined because of its poor solubility in water, due to the larger molecular weight of PCL blocks. The values clearly indicate that CMC of the star-shaped block copolymers decreases with an increase of its hydrophobic character. Hence, the formation of polymeric micelles should be more efficient when the hydrophobic blocks in [PEO(PCL)2] copolymers are of higher molecular weight.

A CMC curve (a) and a TEM micrograph of [PEO25(PCL17)2] triarm block copolymer (b)
The morphology and size of the polymer micelles were investigated by TEM. As seen from the micrograph, (Fig. 5b) the obtained micelles possess a well-defined spherical shape. More than 100 nanoparticles were counted and measured to determine the diameter of the polymeric nanospheres (Fig. 3 in Supplementary Data). It generally varies from 10 to 16 nm with an average size of about 13 nm. In comparison with the linear di- and triblock copolymer counterparts (PEO-b-PCL and PCL-PEO-PCL) [41–43] the micelles formed by [PEO(PCL)2] star-shaped block copolymers have a smaller diameter. Undoubtedly, the formation of polymeric micelles of smaller size and lower CMC values is due to the ability of these copolymers (miktoarm star with double solvophobic tails) to form stable micelles of a lower aggregation number [44].
3.4 Cytotoxicity evaluation
A promising in cell delivery carrier should manifest both sufficient cell penetration efficiency and low cytotoxicity. Therefore the polymer micelles were subjected to MTT assay to determine the cytotoxicity in HepG2 cells. In a functional mitochondria MTT is reduced to formazan by mitochondrial succinate dehydrogenase in complex II (succinate:ubiquinone oxidoreductase complex), which has a crucial role in both the oxidative phosphorylation and tricarboxylic acid cycle [45].
As evident from the cell viability data (Fig. 6), all tested concentrations of the polymeric micelles proved to be free of cytotoxic effects following continuous exposure at 37 °C for 24, 48 and 72 h. There was some statistically insignificant decrease in cell viability (p > 0.05) only at the highest concentration of micellar solution (21.5 μM) for a 24 h exposure. In contrast, cells treated for 48 or 72 h with polymeric solutions (8.6 and 21.5 μM) showed a tendency to increase their viability (Fig. 6). The registered effect may be explained by increased redox activity of mitochondria in the presence of PCL. Similar results were also observed by other authors when culturing L929 fibroblasts on PCL thin films for a short period of time [46] or in the presence of polymeric materials [47].

Viability of HepG2 cells following incubation of micellar solutions at different concentrations for 24, 48 and 72 h. Data are presented as average ± SD of eight independent experiments
The obtained results strongly suggest that the synthesized [PEO(PCL)2] copolymer micelles are non-cytotoxic and they are suitable for in vitro application.
3.5 Cellular uptake and accumulation
Cellular uptake of NR-loaded micelles was demonstrated by light microscopy (Fig. 7a, b) using HepG2 cells. This cell line was chosen because of its important hepatic function and ability to form an adherent cell monolayer in vitro. For the initial 4 h, the uptake of 4.3 μM NR-loaded micelles (Fig. 7a) within the cells was minimal in contrast to that at higher concentration (21.5 μM, Fig. 7b).

Cellular uptake of NR-loaded micelles by HepG2, after incubation for 4 h at 37°C: a 4.3 μM; b 21.5 μM. The arrows show some of the positively stained cells. The images are representative of three experiments, magnification ×200; c time dependent uptake of 21.5 μM [PEO(PCL)2]-NR complex by HepG2 cells. Data are presented as average ± SD of eight independent experiments. *Significantly different from 4 and 24 h uptake, p < 0.001
To further analyze the time dependency of NR-loaded micelle accumulation we incubated the HepG2 cells up to 72 h, followed by an extraction of NR and spectrophotometric quantification at 540 nm. The uptake was observed to increase progressively over time, showing a 1.5 times higher accumulation at the 4th h and another 4.5 times at 72th h compared with NR alone (21.5 μM, Fig. 7c).
The micelle internalization process into the cells can be viewed as a binding/adsorbing process followed by the formation of micelle-containing vesicles that can be internalized by endocytosis [48–50]. Endocytosis is a multi-step complex cellular pathway for the internalization of different macromolecules. To elucidate the internalization mechanism of the NR-loaded micelles by HepG2 cells, various inhibitors were used to study the possible endocytotic mechanisms. Inhibition of cellular uptake was performed in the presence of optimized single inhibitor concentration of CytD (a macropinocytosis inhibitor), CytB (a microtubule inhibitor), CPZ (a clathrin-mediated inhibitor), MβC (a caveolae-mediated inhibitor), and with double combinations. The analysis of the cellular internalization of the NR-loaded micelles in HepG2 was done in comparison with that in the absence of inhibitor as a reference. The results showed that HepG2 cells pretreated with MβC, CytD, and CytD + CPZ internalized a significantly smaller amount of micelles compared with the reference—40, 55, and 37%, respectively. Similarly, treatment with CPZ (75%) or CytB (92%) significantly reduced the amount of internalized micelles. These results indicate that the pathway of cellular uptake is predominantly via macropinocytosis and caveolae mediated endocytosis. Upon endocytosis, internalized nanoparticles become entrapped into an intracellular vesicle. Once the polymers were endocytosed, they were found primarily around the nuclear membrane (Fig. 7 b). We also observed that a large fraction of the incubated micelles were internalized by the cells and remained stable during the cell cycle. To evaluate the release of NR from the NR-loaded micelles we studied its accumulation in the lysosomes under physiological conditions in vitro. It is known that having penetrated the cell membrane by nonionic passive diffusion NR concentrates into the lysosomes [51]. Depending on the concentration, NR can bind electrostatically to the lysosomal matrix and interfere with the lysosomal function leading to cell death. The release profiles (Fig. 8) indicate a sustained manner of NR release from the micelles. About two-thirds of the loaded NR was released within 7 days and no clearly expressed initial burst effect was observed. During the first 24 h of incubation 33% of the NR was released. This could be an indication that the whole amount of NR was incorporated into the polymer micelles. These results also indicate a slow rate of NR release, especially if taking into consideration the short incubation periods and high activity of hepatic lipases present in the HepG2 cells and their participation in the micelles degradation.

Release of NR from NR-loaded micelles in HepG2 cells. The cells were loaded for 24 h with 21.5 μM NR-loaded micelles. The release of NR was followed by its accumulation in the lysosomes of HepG2 cells growing in complete DMEM media for the indicated time, (n = 3)
4 Conclusions
A novel and efficient method for the synthesis of well-defined star-shaped block copolymers based on PEO and PCL has been developed. The operational simplicity, high yield and purity of the products have been demonstrated to be the advantages of the applied synthetic strategy to the previously described ones. The capability of the synthesized amphiphilic [PEO(PCL)2] star-shaped copolymers to form spherical polymeric micelles of a smaller average diameter and CMC values lower than those of their linear counterparts has also been demonstrated. Furthermore, our in vitro studies have proven the obtained polymer micelles to be non-toxic and suitable carriers for in cell delivery.
References
Pitsikalis M, Pispas S, Mays JW, Hadjichristidis N. Nonlinear block copolymer architectures. Adv Polym Sci. 1998;135:1–137.
Vora A, Singh K, Webster DC. A new approach to 3-miktoarm star polymers using a combination of reversible addition-fragmentation chain transfer (RAFT) and ring opening polymerization (ROP) via “Click” chemistry. Polymer. 2009;50:2768–74.
Lele BS, Leroux J-C. Synthesis of novel amphiphilic star-shaped poly(ε-caprolactone)-block-poly(N-(2-hydroxypropyl)methacrylamide) by combination of ring-opening and chain transfer polymerization. Polymer. 2002;43:5595–606.
Kim KH, Cui GH, Lim HJ, Huh J, Ahn CH, Jo WH. Synthesis and micellization of star-shaped poly(ethylene glycol)-block-poly(ε-caprolactone). Macromol Chem Phys. 2004;205:1684–92.
Riva R, Schmeits S, Jérôme C, Jérôme R, Lecomte Ph. Combination of ring-opening polymerization and “click chemistry”: toward functionalization and grafting of poly(ε-caprolactone). Macromolecules. 2007;40:796–803.
Rieger J, Coulembier O, Dubois Ph, Bernaerts KV, Du Prez FE, Jérôme R, Jérôme C. Controlled synthesis of an ABC miktoarm star-shaped copolymer by sequential ring-opening polymerization of ethylene oxide, benzyl beta malolactone, and epsilon-caprolactone. Macromolecules. 2005;38:10650–7.
Van Butsele K, Stoffelbach F, Jérôme R, Jérôme C. Synthesis of a novel amphiphilic and pH sensitive ABC miktoarm star terpolymers. Macromolecules. 2006;39:5652–6.
Petrova S, Riva R, Jérôme C, Lecomte Ph, Mateva R. Controlled synthesis of AB2 amphiphilic triarm star-shaped block copolymers by ring-opening polymerization. Euro Polym J. 2009;45(12):3442–50.
Rempp PF, Lutz PJ. Star-shaped polymers. In: Salamone JC, editor. Polymeric materials encyclopedia, vol. 10. Boca Raton: CRC Press; 1996. p. 7880–5.
Bywater S. Preparation and properties of star-branched polymers. Adv Polym Sci. 1979;30:89–116.
Guo A, Liu G, Tao J. Star polymers and nanospheres from crosslinkable diblock copolymers. Macromolecules. 1996;29:2487–93.
Lapienis G. Star-shaped polymers having PEO arms. Prog Polym Sci. 2009;34:852–92.
Sanda F, Sanada H, Shibasaki Y, Endo T. Star polymer synthesis from ε-caprolactone utilizing polyol/protonic acid initiator. Macromolecules. 2002;35:680–3.
Inoue K. Functional dendrimers, hyperbranched and star polymers. Prog Polym Sci. 2000;25:453–571.
Joziasse CAP, Grablowitz H, Pennings AJ. Starshaped poly[(trimethylene carbonate)-co-(ε-caprolactone)] and its block copolymers with lactide/glycolide: synthesis, characterization and properties. Macromol Chem Phys. 2000;201:107–12.
Choi YK, Bae YH, Kim SW. Protein release from microspheres of star-shaped PEO-PLA block copolymers. Proc Int Symp Control Relat Bioact Mater. 1996;23:349–50.
Kennedy JP, Jacob S. Cationic polymerization astronomy. Synthesis of polymer stars by cationic means. Acc Chem Res. 1998;31:835–41.
Hadjichristidis N, Pitsikalis M, Pispas S, Iatrou H. Polymers with complex architecture by living anionic polymerization. Chem Rev. 2001;101:3747–92.
Matthews OA, Shipway AN, Stoddart JF. Dendrimers: branching out from curiosities into new technologies. Prog Polym Sci. 1998;23:1–56.
Deng M, Chen X, Piaol L, Zhang X, Dai Z, Zing XJ. Synthesis of four-armed poly(ε-caprolactone)-block-poly(ethylene oxide) by diethylzinc catalyst. J Polym Sci Part A Polym Chem. 2004;42:950–9.
Park SY, Han DK, Kim SC. Synthesis and characterization of star-shaped PLLA-PEO block copolymers with temperature-sensitive sol–gel transition behavior. Macromolecules. 2001;34:8821–4.
Kim SH, Han Y-K, Kim YH, Hong SI. Multifunctional initiation of stannous octoate/pentaerythritol for the polymerization of lactide. Makromol Chem. 1992;193:1623–31.
Kim SH, Han Y-K, Ahn K-D, Kim YH, Chang T. Preparation of star-shaped polylactide with pentaerythritol and stannous octoate. Makromol Chem. 1993;194:3229–36.
Gonçalves da Silva AM, Filipe EJM, d’Oliveira JMR, Martinho JMG. Interfacial behavior of poly(styrene)-poly(ethylene oxide) diblock copolymer monolayers at the air-water interface. Hydrophilic Block Chain Length and Temperature Influence. Langmuir. 1996;12:6547–53.
Gan Z, Jim TF, Li M, Yuer Z, Wang S, Wu C. Enzymatic biodegradation of poly(ethylene oxide-b-ε-caprolactone) diblock copolymer and its potential biomedical applications. Macromolecules. 1999;32:590–4.
Kumar N, Ravikumar MNV, Domb AJ. Biodegradable block copolymers. Adv Drug Deliv Rev. 2001;53:23–44.
Ryu J-G, Jeong Y-I, Kim I-S, Lee J-H, Nay J-W, Kim S-H. Clonazepam release from core-shell type nanoparticles of poly(ε-caprolactone)/poly(ethylene glycol)/poly(ε-caprolactone) triblock copolymers. Int J Pharm. 2000;200:231–42.
Van Butsele K, Jérôme R, Jérôme C. Functional amphiphilic and biodegradable copolymers for intravenous vectorisation. Polymer. 2007;48:7431–43.
Mikhail AS, Allen C. Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J Control Release. 2009;138:214–23.
Allen C, Han J, Yu Y, Maysinger D, Eisenberg A. Polycaprolactone-b-poly(ethylene oxide) copolymer micelles as a delivery vehicle for dihydrotestosterone. J Control Release. 2000;63:275–86.
Forresta ML, Wonb Ch-Y, Malickb AW, Kwon GS. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(ε-caprolactone) micelles. J Control Release. 2006;110(2):370–7.
Wang Y, Wang C, Fu S, Liu Q, Dou DY, Lv H, Fan M, Guo G, Luo F, Qian Z. Preparation of tacrolimus loaded micelles based on poly(ɛ-caprolactone)-poly(ethylene glycol)-poly(ɛ-caprolactone). Int J Pharm. 2011;407(1–2):184–9.
Meier MAR, Aerts SNH, Staal BBP, Rasa M, Schubert US. PEO-b-PCL block copolymers: synthesis, detailed characterization, and selected micellar drug encapsulation behavior. Macromol Rapid Commun. 2005;26:1918–24.
Coleman R, Roma MG. Hepatocyte couplets. Biochem Soc Trans. 2000;28:136–40.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63.
Kavros A, Robinson LY, Rimmer S. Effect of dimethylformamide on the ring opening insertion polymerization of ε-caprolactone. J Chem Res (S) 1999;452–3.
Dong PW, Wang XH, Gu YC, Wang YJ, Wang YJ, Gong CY, Luo F, Guo G, Zhao X, Wei YQ, Qian ZY. Self-assembled biodegradable micelles based on star-shaped PCL-b-PEG copolymers for chemotherapeutic drug delivery. Colloids Surf A Physicochem Eng Aspec. 2010;358:128–34.
Van Butsele K, Sibret P, Fustin CA, Gohy JF, Passirani C, Benoit J-P, Jérôme R, Jérôme CJ. Synthesis and pH-dependent micellization of diblock copolymer mixtures. Colloid Interf Sci. 2009;329:235–43.
Allen C, Yu Y, Maysinger D, Eisenberg A. Polycaprolactone-b-poly(ethylene oxide) block copolymer micelles as a novel drug delivery vehicle for neurotrophic agents FK506 and L-685,818. Bioconjug Chem. 1998;9:564–72.
Lu C, Liu L, Guo S-R, Zhang Y, Li Z, Gu J. Micellization and gelation of aqueous solutions of star-shaped PEG-PCL block copolymers consisting of branched 4-arm poly(ethylene glycol) and polycaprolactone blocks. Euro Polym J. 2007;43:1857–65.
Zhang Y, Zhuo R-X. Synthesis and in vitro drug release behavior of amphiphilic triblock copolymer nanoparticles based on poly (ethylene glycol) and polycaprolactone. Biomaterials. 2005;26:6736–42.
Richter A, Olbrich C, Krause M, Hoffmann J, Kissel T. Polymeric micelles for parenteral delivery of Sagopilone: physicochemical characterization, novel formulation approaches and their toxicity assessment in vitro as well as in vivo. Eur J Pharm Biopharm. 2010;75(2):80–9.
Gong CY, Shi S, Wu L, Gou ML, Yin QQ, Guo QF, Dong PW, Zhang F, Luo F, Zhao X, Wei YQ, Qian ZY. Biodegradable in situ gel-forming controlled drug delivery system based on thermosensitive PCL-PEG-PCL hydrogel. Part 2: Sol–gel–sol transition and drug delivery behavior. Acta Biomater. 2009;5:3358–70.
Lin C-M, Chen Y-Z, Sheng Y-J, Tsao H-K. Effects of macromolecular architecture on the micellization behavior of complex block copolymers. React Funct Polym. 2009;69:539–45.
Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–69.
Serrano MC, Pagani R, Vallet-Regí M, Peña J, Rámila A, Izquierdo I, Portolés MT. In vitro biocompatibility assessment of poly(ε-caprolactone) films using L929 mouse fibroblasts. Biomaterials. 2004;25(25):5603–11.
Ignatius AA, Claes LE. In vitro biocompatibility of bioresorbable polymers: poly(l, dl-lactide) and poly(l-lactide-coglycolide). Biomaterials. 1996;17:831–9.
Foster KA, Yazdanian M, Audus KL. Microparticulate uptake mechanisms of in vitro cell culture models of the respiratory epithelium. J Pharm Pharmacol. 2001;53:57–66.
Jule E, Nagasaki Y, Kataoka K. Lactose-installed poly(ethylene glycol)-poly(d,l-lactide) block copolymer micelles exhibit fast-rate binding and high affinity toward a protein bed simulating a cell surface. A surface plasmon resonance study. Bioconjug Chem. 2003;14:177–86.
Allen C, Yu Y, Eisenberg A, Maysinger D. Cellular internalization of PCL20-b-PEO44 block copolymer micelles. Biochim Biophys Acta. 1999;1421:32–8.
Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3(7):1125–31.
Acknowledgments
The authors thank to Solvay Chemicals (sector-Solvay UK Caprolactones) for providing the excellent quality material (ε-caprolactone, CAPA®). This article has been compiled with the financial support of Project BG051PO 001-3.3.04/58 of the “Human Resources Development” Operational Programme, co-financed by the European Union through the European Social Fund. The whole responsibility for the article contents lies with the Beneficiary and under no circumstances should this article be regarded as representing the official position of the European Union and the Contract Body.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Petrova, S., Kolev, I., Miloshev, S. et al. Synthesis of amphiphilic [PEO(PCL)2] triarm star-shaped block copolymers: a promising system for in cell delivery. J Mater Sci: Mater Med 23, 1225–1234 (2012). https://doi.org/10.1007/s10856-012-4592-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-012-4592-8