
Abstract
Design and development of nanoparticle-based drug delivery systems (DDS) have always been extremely challenging due to unacceptable leakage of drug or unsatisfactory release in the lesion. Herein, we presented an effective approach for facile preparation of DDS by imprinting doxorubicin (DOX) via miniemulsion polymerization. In order to investigate the release of DOX in this system, the cumulative release of DOX by molecularly imprinted polymers (GMIPs) and non-imprinted polymers (GNIPs) was compared. The results revealed that the DOX’s leakage of GMIPs was more moderate than that of GNIPs. Moreover, after fitting the release curves with several mathematical models, it was found that the release of DOX from the GMIPs can be partially fitted with zero-order model (R2 = 0.929) which implied that molecular imprinting techniques for drug loading could reduce the common ‘burst effect.’ In addition, excellent release of DOX with controllable property was achieved by switching photothermal effect of graphene oxide quantum dots which were doped in GMIPs as the near-infrared light (NIR) window. Thus, it would be anticipated that the novel drug-loaded GMIPs combining with inductive NIR heating would be promising to be applied in the synergy of chemotherapy and thermotherapy for cancer therapy.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Drug delivery system (DDS) is the strategy that transports drugs into the specific site to achieve the desired therapeutic effect [1]. To maximize the safety and efficacy of drugs, DDS should be able to prevent the premature leakage of drugs to lessen harm of the normal cells and maintain their concentration in the therapeutic window [2,3,4]. Therefore, the development of drug loading with high loading efficacy and little leakage has become more urgent [5, 6]. Generally, drug loading involved physical adsorption [7], chemical conjugation [8] and solvent evaporation [9]. Nevertheless, the majority of studies focus on drug release rather than drug loading [10], which has slowed down the development of DDS.
Molecular imprinted polymers (MIPs) are cross-linked polymers that exhibit specific binding sites for the target molecule [11, 12]. The most common strategy for the preparation of MIPs only needs one-step self-assembly of functional monomer with template molecule via non-covalent interactions and then the polymerization with the help of cross-linking agent as well as initiator, which is effortless and well established [13,14,15,16]. On the basis of their unique properties and multiple forces generated from imprinting, MIPs seem to be perfectly designed for drug loading [1, 17]. For example, Zhang et al. [10] reported a surface molecular imprinting polymer using mesoporous silica nanoparticles as substrate with loading efficiency of 70 ± 8% for DOX. In another work, paclitaxel molecularly imprinted polymer-PEG-folate nanoparticles were designed, which showed high drug loading and encapsulation efficiency of 15.6 ± 0.8 and 100%, respectively [18]. Moreover, the release profile showed that the burst release within 24 h was only 11.2% (pH = 5) and 6.7% (pH = 6) [18]. These researches clearly indicated that MIPs possess great potential for the drug loading with high loading efficacy and little leakage.
Traditionally, MIPs were prepared by bulk polymerization with the following crushing and sieving to obtain polymer beads [19]. However, several methods can be used to synthesize polymer micro- and nanoparticles directly, thus could avoid the time- and labor-consuming process of crush sieving. Miniemulsion polymerization is one of the desirable methods, and it could incorporate drug and quantum dots into the polymerization for functional polymer particles [20]. However, the elution of surfactants after polymerization is not easy, and the adsorbed and absorbed surfactants desorb with time, possibly leading to physical discomfort [21]. Fortunately, polymerizable surfactants perfectly solve the above problems. In addition, it was reported that the imidazolium-based ionic liquids surfactants with alkyl substitution of C-12 chain length were found to be effective against microbes [22] and shown very low cytotoxicity in most of the cases [23,24,25]. Moreover, due to the difference in biochemical properties of microbes, normal cells and tumor cells, the positive charge that is carried out by the cationic surfactant has passive targeting ability in tumor tissue and selective killing to microbe [26, 27]. It is gratified that polymer materials used in anticancer drug delivery system could satisfy both antimicrobial and biocompatibility properties [28]. Therefore, utilizing polymerizable imidazolium-based ionic liquids as surfactants for MIPs is very promising.
It should be noted that DDS not only prevents drug leakage but also requires immediate release of the drug at a specific site [29]. Generally, the release of DDS was triggered by distinct external stimulation, such as pH [4, 10, 30, 31], temperature [30, 31], magnetism [30], redox-responsive [32] or NIR [33]. In recent years, more and more insights have been gained on cancer therapy through the combining the synergistic effect of chemotherapy with NIR photothermal therapy [33, 34]. Graphene quantum dots (GQDs) are greatly desirable for the development of DDS because of their strong NIR absorbance, high photothermal conversion efficiency and excellent thermal conductivity in vivo [35]. Simultaneously, their high surface area and various surface functional groups make it possible to load drugs [36]. Moreover, the combination of molecular imprinting technology and quantum dots has received widespread attention in recent years, which provides favorable references for our work [37,38,39].
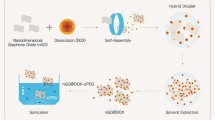
Based on the above-mentioned factors, the present study describes the synthesis of molecular imprinted polymers doped GQDs (GMIPs) and their application in instantaneous release of the anticancer drug doxorubicin (Scheme 1). Specifically, a antibacterial cationic polymerizable surfactant, 1-vinyl-3-dodecylimidazolium bromide (ViDoIm+Br−) was prepared and used as the emulsifier in miniemulsion polymerization for the preparation of GMIPs. During the polymerization, DOX and GQDs were encapsulated in the polymer to achieve loading and photothermal conversion in the selection of NIR window. Valuable properties of the obtained DDS include: safe delivery to avoid premature drug release, precise treatment in the lesion site because of the triggering mechanisms of NIR, and antibacterial surface to meet the requirements of anticancer drug delivery system.

Schematic diagram of GMIPs synthesis and DOX release by an ex vivo
Experimental section
Chemicals and materials
GQDs were obtained from Nanjing XFNANO Materials Tech Co., Ltd. 1-Bromododecane, 1-vinylimidazole, doxorubicin hydrochloride (DOX, 98%), dodecyltrimethylammonium bromide (DTAB), ethylene glycol dimethacrylate (EGDMA, 98%), methyl methacrylate (MMA, 99%), cetyl alcohol (CA), 2,2-azobisisobutyronitrile (AIBN) were purchased from Shanghai Macklin Biochemical Co., Ltd. Deionized water was used in all experiments.
Synthesis of ViDoIm+Br−
The product, ViDoIm+Br−, was prepared referencing the procedures described in the literature [23]. Under vigorous stirring, 5.99 mL (0.025 mol) of 1-bromododecane were added dropwise to 2.27 mL (0.025 mol) of 1-vinylimidazole in a 50 mL round bottom flask with one necked. The mixture was reacted at 45 °C for 72 h. The resulting white–yellow solid was washed several times with ethyl acetate. And then, the product was filtered and dried in a vacuum oven until constant weight.
Determination the micellar properties of surfactants
Conductivity method is an easy method for determining the critical micelle concentration (CMC) of surfactants [40]. ViDoIm+Br− was added to pure water, and a series of precise concentrations of surfactants in aqueous solution were formulated, respectively. The solution was stirred for 1 min prior to the conductivity measurement.
Synthesis of polymer microspheres via miniemulsion polymerization
All the components required for the preparation of GNIPs were divided into two parts [41]. One was an aqueous phase, which included 50 mL deionized water, 100 mg of ViDoIm+Br− and 6 mg of GQDs. The other was oil phase, which composed of 400 μL of EGDMA, 200 μL of MMA, 10 mg of CA. The oil phase without initiator was mixed by stirring for 10 min and was ultrasonicated in an ice bath for 10 min to obtain a homogenous dispersion. Then, 10 mg of AIBN was dissolved in the oil phase. The oil phase containing the initiator was slowly added to the aqueous phase and pre-emulsifying the O/W mixture by magnetic stirring for 30 min. Then, the O/W mixture was ultrasonicated using a ultrasonicator (KQ-200KDE Sonifier, Kunshan Shumei, China) in an ice bath for 20 min (input power 200 W, frequency 40KHZ). The obtained miniemulsion was stirred for 30 min to homogenize the system. Afterward, the miniemulsion was poured into 100 mL three-necked glass reactor equipped with condenser and mechanical stirrer in an oil bath and was carried out at 70 °C for 4 h under stirring rate 300 rpm. After polymerization, the product was demulsified and purified from surfactant by centrifugation (at 5000 rpm for 5 min) and redispersion in anhydrous ethanol for four times. Then, the microspheres were washed thoroughly with distilled water. Finally, GNIPs was obtained after freeze-drying for 24 h. The same protocol was followed for preparation of GMIPs, but adding 3 mg DOX to the oil phase early for pre-assembling with the monomer.
To highlight the effectiveness of surfactants ViDoIm+Br− designed in our work, we selected DTAB whose chemical structure was similar to ViDoIm+Br− to prepare corresponding GNIPs-DTAB as references. The protocol of GNIPs was followed for preparation of GNIPs-DTAB, but DTAB was substituted for ViDoIm+Br− in the aqueous phase. The detailed composition is shown in Table 1.
Antibacterial tests
Antibacterial tests were performed to examine the antimicrobial properties of GNIPs-DTAB, GNIPs and unloaded GMIPs (after template removal) microspheres against the Gram-negative bacterium of E. coli and the Gram-positive bacterium of S. aureus. Nutrient agar medium was prepared by adding agar (20.0 g) into the solution (peptone 10.0 g, yeast 5.0 g, sodium chloride 10.0 g, and distilled water 1000 mL) with pH of 7.4. The agar medium was sterilized in a conical flask at 121 °C for 30 min under a pressure of 0.1 MPa. This medium was then transferred into sterilized Petri dishes. The culture of the microorganism was diluted by sterile distilled water to approximately 106 CFU/mL. After solidification of the medium, the mixture of microorganism culture and 2.5 mg/mL microspheres solution was coated on the solid surface of the medium by spread plate method. The colonies were observed after incubating bacteria strains at 37 °C for 24 h.
Determination of the drug loading content and entrapment efficiency
The GMIPs were heated with aqueous solution at 60 °C for two days to remove the template. For comparison, GNIPs were handled by the same operation. After freeze-drying, the intact DOX was then adsorbed as the loading drug. Specifically, 10 mL of 1 mg/mL DOX solution (PBS buffer, pH 7.4) was added to 100 mg of GMIPs or GNIPs, and the mixture was kept shaking for 24 h. Finally, the content of DOX in the supernatant was measured by a UV–visible spectrophotometer. The drug loading content and entrapment efficiency were determined by the following equations:
Drug release behaviors of DOX in vitro
In vitro release experiments were performed in PBS buffer at pH values of 7.4. In detail, 200 mg of GMIPs and GNIPs nanoparticles was placed in a dialysis bag with a molecular weight cutoff of 3500 Da, respectively. In the instantaneous release experiment, the GMIPs solution was additionally exposed to the 808-nm laser at a power density of 2 W/cm2. Samples (3 mL) were periodically collected, and the removed liquid is returned to the original system after the test. The samples were analyzed with a UV–visible spectrophotometer to measure the amount of released DOX. The drug cumulative release at time t (Rt) was determined by the following equations:
In order to effectively simulate and predict drug release behavior, drug release data were studied using zero-order (Eq. 4), first-order (Eq. 5), Higuchi (Eq. 6), and Hixson–Crowell (Eq. 7) mathematical models.
where K0, K1, KH, and KHC are the release constants of the respective equations.
Characterization
In order to elucidate the structure of prepared polymerizable ionic liquid, nuclear magnetic resonance (NMR, Bruker Avance 400 MHz, Bruker, Germany) using CDCl3 as solvent was performed. The conductivity measurements of surfactant solutions were taken at 25 °C using a conductometer (DDS-307, Leici, China), and the average of three measurements was taken. Transmission electron microscopy (TEM, FEI Talos F200X, FEI, USA) was employed to obtain the morphologies and size of the prepared microspheres. FTIR spectra in the region of 4000–400 cm−1 were recorded by the KBr pellet method with Fourier transform infrared spectrometer (FTIR, TENSOR27, Bruker, Germany). Chemical surface characterization was performed by X-ray photoelectron spectrometry (XPS, Kratos Axis Ultra DLD, Kratos, UK). Zeta potential and dynamic light scattering (DLS) measurements of the prepared microspheres were performed using a Zetasizer Nano detector (Zeta, ZEN3690, Malven, Britain). Thermal data were obtained by a thermogravimetric analyzer (TGA, SDT Q6000, USA) under the temperature range 20–800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
Results and discussion
Synthesis of ViDoIm+Br−
Here, the ionic liquid was not only used as an emulsifier for miniemulsion polymerization, but also capable of participating in the polymerization of GNIPs and GMIPs. Therefore, the ionic liquid needs to satisfy two essential conditions. First, it must be the same like traditional surfactant, consisting both of hydrophilic and lipophilic parts. Second, the resultant surfactant should have the polymerizable double bond. Hence, we synthesized ViDoIm+Br−, which consisted of a hydrophilic imidazole group with a double bond and a hydrophobic dodecyl chain. As shown in Fig. 1, H-NMR of the ViDoIm+Br− was performed to verify the synthesis of the anticipated products. ViDoIm+Br−: 1H-NMR (CDCl3, 400 MHz, ppm): δ10.86 (s, 1H, H1), 7.98 (d, 1H, H2), 7.63 (d, 1H, H3), 7.49 (t, 1H, H4), 6.03 (d, 1H, H5), 5.35 (d, 1H, H6), 4.40 (t, 2H, H7), 1.93 (m, 2H, H8), 1.23 (m, 18H, H9), 0.86 (t, 3H, H10).

1H-NMR spectrum of ViDoIm+Br− in CDCl3
The micellar properties of surfactants ViDoIm+Br−
We anticipated that ViDoIm+Br− would exhibit an interfacial and aggregation behavior which was analogous to the conventional cationic surfactants. Figure 2 shows the change of the conductivity values (δ) with the increase in the surfactant concentration. The breakpoint of two straight lines gave the CMC value of ViDoIm+Br−. Below CMC value, the δ value rose with an increase in surfactant concentration due to the free surfactant ions in the medium. The surfactant molecules began self-aggregating to form micelles when the surfactant concentrations came to the intersection point. The conductivity increased slowly at the concentrations above the CMC because the micelle mobility was lower than the free surfactant ions. The experimental result showed that the CMC value of ViDoIm+Br− was 4.1 × 10−3 M and ViDoIm+Br− would exhibit rather good surface activity.

Variation in the conductivity with ViDoIm+Br− concentration in aqueous solution
Morphology and characterization of polymer microspheres
The morphology and size of polymer microspheres were characterized by TEM. In Fig. 3b, c, the GNIPs and GMIPs synthesized using ViDoIm+Br− as surfactant showed a comparatively broad distribution of sizes ranging from 80 to 600 nm. A few reports have declared that the particles which as the delivery of pharmacologically active agents less than 5 µm are relatively safe, because the size range has undemanding access to absorbing and transporting into the bloodstream [42]. Therefore, this system would be suitable for clinical application in some extent. GNIPs-DTAB synthesized using DTAB as surfactant was basically the same as GNIPs, except for bigger size (Fig. 3a). In addition, the DLS measurement results were consistent with TEM and the size of the three microspheres was not uniform, which indicated that the microspheres prepared via miniemulsion polymerization method using considerable amounts of cross-linkers had a polydisperse diameter.

TEM images and size distributions as measured by DLS of GNIPs-DTAB (a), GNIPs (b), and GMIPs (c)
To further ascertain the properties of the polymer microspheres, other characterizations were employed. The results of XPS characterization are shown in Fig. 4. The only source of N elements was from the surfactants due to the absence of N in MMA and EGDMA. Since most of DTAB was washed away after rinsing with water, the characteristic peaks of N 1s were not observed in the wide scan spectra and high magnification of N 1s of GNIPs-DTAB (Fig. 4a, b). However, N 1s of GNIPs still could not be detected in XPS wide scan (Fig. 4a), which was due to the complete concealment of N 1s by long alkyl chains of the polymerizable surfactant [43]. Fortunately, there was a characteristic peak in GNIPs microspheres after high magnification which was assigned to the nitrogen (N 1s) of the imidazole groups (Fig. 4b). As shown in Table 2, the N content in GNIPs was 0.44%, which completed the transition from scratch. Such a significant change in N content indicated that more polymerizable surfactants eventually stayed on the surface of the GNIPs. The content of N element in the GMIPs was similar to that of GNIPs. In order to indicate the successful incorporation of functional monomers and cross-linkers into GNIPs-DTAB, GNIPs and GMIPs, FTIR characterization is shown in Fig. 5. The peak at 1732 cm−1 was assigned to the C=O stretching of EGDMA, and the peaks around 1257 and 1152 cm−1 were ascribed to C–O–C stretching. Note that the characteristic adsorption peaks of poly (MMA) in poly (MMA-co-EGDMA) microspheres were not discernible due to their overlap with those of poly (EGDMA). Moreover, the C=C functional groups at around 1634 cm−1 were found [23].

XPS wide scan spectra (a) and high magnification of N 1s (b) of GNIPs-DTAB, GNIPs, and GMIPs

FTIR spectra of GNIPs-DTAB, GNIPs and GMIPs
Zeta potential of different microspheres is presented in Table 2. For GNIPs, zeta potentials remained positive (43.09 mV), demonstrating that most of the surfactants involved in the polymerization still existed on the surface after washing. Although DTAB was not detected in the high magnification of N 1s, GNIPs-DTAB surface charge still showed to the relatively low positive zeta potential value (8.14 mV) after washing, indicating that although most DTAB had been ‘washed off’ (desorbed) from the surface, a few remained [44]. The zeta potential of GMIPs (36.36 mV) was slightly lower than GNIPs. The reasonable explanation might be that the interaction sites between the microspheres and GQDs increase after the addition of the template DOX, causing more GQDs to be attracted and more positive charges to be neutralized.
NIR photothermal conversion
We irradiated these polymer microspheres solutions (6 mg/mL) with an 808-nm laser for 40 min at a laser power of 2 W/cm2. As shown in Fig. 6, the temperature of all solutions increased rapidly under irradiation within five minutes and then leveled off at 38, 40, and 43 °C for GNIPs-DTAB, GNIPs, and GMIPs, respectively. Variations in temperature might be related to the density of GQDs. GQDs with negatively charged because of the presence of hydroxyl and carboxyl groups might show electrostatic interactions with the positively charged imidazole ionic liquids. Therefore, GNIPs and GMIPs showed a higher temperature than GNIPs-DTAB. Zeta potential data indicated that GQDs in GMIPs was more than GNIPs, so the temperature of GMIPs was highest. In theory, protein deformation and DNA damage might occur when the local temperature of the tumor exceeded 42 °C, which would directly kill the cancer cells [45]. Excitingly, DDS performed enhanced permeation retention effect (EPR) under tumor microenvironments, which benefited the nanospheres accumulation [46], possibly making higher local temperature with NIR. Therefore, this system would have the possibility of clinical hyperthermia which will not be discussed in this work.

Thermal images of GNIPs-DTAB, GNIPs, and GMIPs solution, respectively, after 40 min (a); temperature change curves of GNIPs-DTAB, GNIPs, and GMIPs solution exposed to the 808-nm laser at a power density of 2 W/cm2 (b)
Antimicrobial activity
Because of the severe decline in body immunity, cancer patients are susceptible to bacterial infection after chemotherapy. Inflammatory cells can also affect cancer metastasis. Thus, it is necessary to avoid infection during cancer treatment [47, 48]. The aim of this study was to synthesize a novel multi-potent smart antibacterial nanocarrier for anticancer drug delivery purposes. Therefore, antimicrobial tests of GNIPs were carried out and the results were compared with GNIPs-DTAB (Fig. 7). As shown in the horizontal row of GNIPs, after co-culture with GNIPs for 24 h, the viable colonies of S. aureus and E. coli decreased sharply. The quaternary ammonium compounds such as imidazolium-based ionic liquids exhibit a strong antimicrobial activity [49]. The antimicrobial activity was mainly due to the interaction of the ammonium salt with the cell membrane of microorganisms [50]. On the contrary, the horizontal row of GNIPs-DTAB showed relatively poor antimicrobial activity. It was due to the fact that the content of DTAB was too low to exhibit significant antibacterial properties, although DTAB was also a quaternary ammonium salt compound [51]. In addition, the electrostatic attraction between positively charged nanoparticles and negatively charged bacterial cells was shown to be an important aspect with regard to the antimicrobial activity [52, 53]. The zeta potential of GNIPs (43.09 mV) was slightly higher than GMIPs (36.36 mV), so the results show that the antimicrobial effect of GNIPs was better than that of unloaded GMIPs. But overall, the antimicrobial surface caused by ionic liquids would allow polymeric materials to be better used in anticancer drug delivery systems. So far, the characterization of materials has been completed. Next, we have begun to focus on the properties of GNIPs and GMIPs as drug delivery systems.

Antimicrobial activities of GNIPs-DTAB, GNIPs and GMIPs against E.coli (a) and S. aureus (b)
Characterization of drug loading
The DOX loading to GMIPs had been calculated as high as 7.08%, corresponding to the encapsulation efficiency of 70.8%. Compared to other DDS in Table 3, the drug loading content and encapsulation efficiency of the new materials in this work were similar to or even better than that of the general polymer drug delivery system, although there was still a certain gap compared with nanogels and mesoporous materials. The drug loading depends on two reasons.
First, the GQDs have shown extraordinary potential in the field of drug delivery because of their six-membered carbon ring structure, which could allow π–π and hydrophobic interactions with aromatic rings of DOX [60]. Figure 8 illustrates the UV–Vis spectra of GQDs, free DOX, and DOX-GQDs. Free DOX displayed absorption at 233.6, 254.0, and 480.5 nm (black line), and the peak was slightly redshifted from 480.5 to 485.0 nm after DOX adsorbed on the surface of GQDs (green line), indicating DOX loaded to GQDs successfully. In addition, GQDs as a new class of two-dimensional materials with large specific surface area [61], could not only improve the affinity and sensitivity of the template drug molecules, but also improve the drug loading of the existing MIP-based DDS.

UV-Vis absorption spectra for free DOX, GQDs with and without bound DOX
Second, there might be various kinds of interactions between template drugs and imprinting sites of polymers in aqueous medium [1]. To illustrate the interaction between DOX and MMA, the pore size distribution of GNIPs and GMIPs was examined by the nitrogen adsorption–desorption isotherms and the corresponding BJH pore size distributions curves. The results are shown in Fig. 9 that both samples showed obvious hysteresis loop because of the existence of pores [62]. The porous structure of polymers resulted from a phase separation between the polymer and cetyl alcohol as porogen during the polymerization [63]. Moreover, the pore diameter of GNIPs and GMIPs was measured to be 1.93 and 2.53 nm, respectively. The larger pore size of GMIPs was derived from the intermolecular forces between DOX and functional monomers MMA. In addition, many researchers have also confirmed that the molecular imprinting technique was developed based on interaction between template drug molecules and suitable functional monomers [1, 10, 58].

N2 adsorption–desorption isotherms (a) and pore size distribution (b) for GNIPs and GMIPs
Figure 10 illustrates the results of thermogravimetric analysis of loaded GMIPs (before template removal), unloaded GMIPs (after template removal), GNIPs and DOX. For all polymer microspheres, similar results were obtained. An initial mass loss was observed below 300 °C due to continuous removal of solvents and water that was enclosed in the cross-linked polymers. The dramatic weight loss was observed at approximately 300–450 °C which was connected with the decomposition of the polymers. However, there were still differences in the details. At 220 °C, a relatively significant mass change was only observed in loaded GMIPs curve, corresponding to the weight loss of the DOX. In addition, the mass loss of 300–450 °C was slightly lower for the loaded GMIPs compared to the two other polymers. To make this discussion more concrete, the plots were made from the same starting weight (%) at 300 °C (Fig. 10b). The final platform of loaded GMIPs was slightly higher, which clearly indicated that more additional non-volatile material was present in the residue. Because DOX was not fully decomposed into volatile material under nitrogen atmosphere [1], this result indicated that DOX was imprinted in loaded GMIPs’ structure. Therefore, the coverage and drug load of GNIPs were lower than the corresponding values of GMIPs, which were 4.00% and 39.98%, respectively.

TGA results of drug-loaded GMIPs, drug-unloaded GMIPs, and GNIPs. Temperature range: 20–800 °C (a); 300–800 °C (b)
In addition, the cumulative release of GNIPs and GMIPs over time also demonstrated that molecular imprinting techniques provided multiple forces for drug loading. The typically high loading efficiency of drug molecules indicated high potential of the GMIPs system as nanocarrier.
Drug release behaviors of DOX in vitro
A desirable property of MIPs is the ability to desorb the template in a controlled manner for their application in drug delivery systems [64]. To demonstrate this potential, the kinetic release curve of the relation between the cumulative release of DOX and release time in the solution is shown in Fig. 11a. It can be seen that the DOX released from GNIPs was characterized by an initial rapid release and then a step of slower release. More than 23.34% of the DOX was released from GNIPs after 60 min. In the case of GMIPs, the release of DOX was slower, particularly between 10 and 60 min and less than 14.21% were released after 180 min. These results could be explained by the fact that most of the DOX loading on GNIPs came from weak and non-specific adsorption, as can be seen from the rapid release. DOX-GMIPs interactions dominated the release profile of GMIPs because DOX was incorporated into highly specific cavities in the nanoparticles where specific and cooperative retention mechanisms took place, causing slower release. The lower diffusion rate of DOX from GMIPs indicated that the specific binding characteristic of these systems can provide a useful means of sustaining the delivery profile and prevent drugs from leaking during delivery.

Kinetic release curve of DOX from GNIPs to GMIPs (a); The cumulative release of DOX from GMIPs under or without the irradiation of 808-nm laser (2 W/cm2) for different time (b)
It was well known that ideal drug release was carried out by controllably switching on or off the drug supply in order to maintain the active drug at the appropriate concentration in vivo for the required time to produce therapeutic effect [33]. To realize the on or off release of DOX, GMIPs aqueous dispersions were continuously irradiated by an 808-nm laser with a power density of 2 W/cm2 for different times (Fig. 11b). For comparison, the release of DOX from GMIPs nanocomposites (without NIR) was also recorded under otherwise identical conditions. The cumulative release of DOX without NIR was 4.05% at 30 min, and then increased slowly with time. When the NIR fell into the particles only for 30 min, the temperature was still in the rising stage at this moment, which was not only unfavorable for the change of binding energy between GODs and DOX [65], but also for the acceleration of thermal motion of drug molecules. That is why the amount of drug release low at 4.86% until 30 min. But then, the cumulative release of DOX increased almost linearly to 26.02% at 90 min, exhibited a slow increase to 36.54% (at 180 min). This controllable release results showed the fact that GQDs in the GMIPs microspheres converted the 808-nm laser energy to heat and then conferred the temperature elevation, leading to change of the binding energy between GQDs and DOX [65]. Moreover, the release kinetics of 34, 37, 40 and 43 °C (Fig. 12) expectedly revealed the important influence of temperature changes on the drug release performance. Compared to lower temperatures, higher temperatures could lead to increased release rates and cumulative release. Simply raising the temperature was also conducive to drug release, so more DOX were released from GMIPs with NIR switching on.

The effect of temperature on drug release behavior of GMIPs
Several mathematical models were utilized for analyzing the DOX release. The values of corresponding release constants k and correlation coefficients (R2) are collected in Table 4. For GNIPs, GMIPs, GMIPs (NIR), the release of DOX could be nicely fitted with first-order model (R2 = 0.924, 0.939, and 0.959, respectively). This implied that drugs were released relatively rapidly at first, but then the release significantly decelerates [66]. The release of DOX from the GMIPs can be partially fitted with zero-order model (R2 = 0.929), whereas for GNIPs, the R2 values are relatively low (R2 = 0.888). It was reported that zero-order release systems could minimize the side effect of the drug, as they allow the delivery of drugs without the common ‘burst effect’ in which a significant amount of drugs was released in the initial stage of the process [67]. Although the obtained R2 value of GMIPs (NIR) (R2 = 0.934) was similar to GMIPs, noticeable differences in Ko values indicated that the release rate of DOX from GMIPs (NIR) was significantly faster than GMIPs. Compared with Hixson–Crowell mathematical models, the release of DOX from GNIPs and GMIPs was perfectly fitted with the Higuchi model (R2 = 0.974 and 0.973, respectively). It can be assumed that the release mechanism of two materials was mostly based on Fickian diffusion. The R2 value of Higuchi model by GMIPs (NIR) was relatively low, which indicates that the release mechanism was different from that of GMIPs due to photothermal effect.
Conclusions
Here, we successfully prepared a polymer microsphere doped with GQDs via miniemulsion polymerization using a polymerizable ionic liquid ViDoIm+Br− as emulsifier and a photothermally triggered molecular imprinted DDS was designed by adding the template DOX. The GMIPs dramatically enhanced DOX loading efficiency and weakened drug leakage rate in comparison with GNIPs because of multiple forces provided by molecular imprinting techniques. Importantly, the effective releasing of loaded DOX from GMIPs was controlled by NIR irradiation due to the photothermal effect of GQDs. This work has provided an idea for designing novel drug delivery systems, which allows the drug to reach the therapeutic effect only at suitable location. We believe that the combination of molecular imprinting techniques and photothermally controllable drug delivery systems would exhibit great potential application in nanomedicine fields.
References
Cegłowski M, Hoogenboom R (2018) Molecularly imprinted poly(2-oxazoline) based on cross-Linking by direct amidation of methyl ester side chains. Macromolecules 51:6468–6475
Hoag H (2014) Brain food. Nature 510:6–7
Kalaydina RV, Komal B, Qorri B, Decarlo A, Szewczuk MR (2018) Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int J Nanomed 13:4727–4745
Hou SL, Chen SS, Dong Y, Gao S, Zhu BS, Lu QH (2018) Biodegradable cyclomatrix polyphosphazene nanoparticles: a novel pH-responsive drug self-framed delivery system. ACS Appl Mater Inter 10:25983–25993
Tibbitt MW, Dahlman JE, Langer R (2016) Emerging frontiers in drug delivery. J Am Chem Soc 138:704–717
Jeon H, Kim MS, Hong SK, Cho SJ, Lim G (2018) Tensile strain-controlled drug delivery system based on a cracked metal structure. Sensor Actuat B-Chem 270:64–71
Janićijević J, Krajišnik D, Calija B, Dobričić V, Daković A, Krstić J, Marković M, Milić J (2014) Inorganically modified diatomite as a potential prolonged-release drug carrier. Mat Sci Eng C-Mater 42:412–420
Pramod PS, Shah R, Jayakannan M (2015) Dual stimuli polysaccharide nanovesicles for conjugated and physically loaded doxorubicin delivery in breast cancer cells. Nanoscale 7:6636–6652
Preisig D, Haid D, Varum FJO, Bravo R, Alles R, Huwyler J, Puchkov M (2014) Drug loading into porous calcium carbonate microparticles by solvent evaporation. Eur J Pharm Biopharm 87:548–558
Zhang KL, Guan XJ, Qiu YX, Wang DD, Zhang XY, Zhang HX (2016) A pH/glutathione double responsive drug delivery system using molecular imprint technique for drug loading. Appl Surf Sci 389:1208–1213
Pakulska MM, Miersch S, Shoichet MS (2016) Designer protein delivery: from natural to engineered affinity-controlled release systems. Science 351:aac4750. https://doi.org/10.1126/science.aac4750
Ding SC, Hu XL, Guan P, Zhang N, Li J, Gao XM, Zhang XY, Ding XL, Du CB (2017) Preparation of surface-imprinted microspheres using ionic liquids as novel cross-linker for recognizing an immunostimulating peptide. J Mater Sci 52:8027–8040
Li DM, He QH, He YH, Xin MG, Zhang YL, Shen ZY (2017) Molecular imprinting sensor based on quantum weak measurement. Biosens Bioelectron 94:328–334
Zhang X, Yang S, Jiang R, Sun LQ, Pang SP, Luo AQ (2018) Fluorescent molecularly imprinted membranes as biosensor for the detection of target protein. Sensor Actuat B-Chem 254:1078–1086
Mourão CA, Bokeloh F, Xu JJ, Prost E, Duma L, Merlier F, Bueno SMA, Haupt K, Bui BTS (2017) Dual-oriented solid-phase molecular imprinting: toward selective artificial receptors for recognition of nucleotides in water. Macromolecules 50:7484–7490
Piletska E, Yawer H, Canfarotta F, Moczko E, Kempisty KS, Piletsky SS, Guerreiro A, Whitcombe MJ, Piletsky SA (2017) Biomimetic silica nanoparticles prepared by a combination of solid-phase imprinting and Ostwald ripening. Sci Rep. 7:11537. https://doi.org/10.1038/s41598-017-12007-0
Zaidi SA (2014) Molecular imprinted polymers as drug delivery vehicles. Drug Deliv 23:2262–2271
Manesh ME, Darvishi B, Ishkuh FA, Shahmoradi E, Mohammadi A, Javanbakht M, Dinarvand R, Atyabi F (2016) Paclitaxel molecularly imprinted polymer-PEG-folate nanoparticles for targeting anticancer delivery: characterization and cellular cytotoxicity. Mat Sci Eng C 62:626–633
Schillemans JP, Nostrum CFV (2006) Molecularly imprinted polymer particles: synthetic receptors for future medicine. Nanomedicine 1:437–447
Wang ZH, Qiu T, Guo LH, Ye J, He LF, Li XY (2018) The synthesis of molecular recognition polymer particles via miniemulsion polymerization. React Funct Polym 126:1–8
Bengani L, Scheiffele G, Chauhan A (2015) Incorporation of polymerizable surfactants in hydroxyethyl methacrylate lenses for improving wettability and lubricity. J Colloid Interf Sci 445:60–68
Fang C, Kong LL, Ge Q, Zhang W, Zhou XJ, Zhang L, Wang XP (2019) Antibacterial activities of N-alkyl imidazolium-based poly(ionic liquid) nanoparticles. Polym Chem-UK 10:209–218
Zhang N, Hu XL, Guan P, Du CB, Li J, Qian LW, Zhang XY, Ding SC, Li BP (2017) Preparation of protein imprinted microspheres using amphiphilic ionic liquid as stabilizer and emulsifier via, miniemulsion polymerization. Chem Eng J 317:356–367
Guo JN, Xu QM, Zheng ZQ, Zhou SB, Mao HL, Wang B, Yan F (2015) Intrinsically antibacterial poly(ionic liquid) membranes: the synergistic effect of anions. ACS Macro Lett 4:1094–1098
Ding SC, Li ZL, Cheng Y, Du CB, Gao JF, Zhang YW, Zhang N, Li ZT, Chang NH, Hu XL (2018) Enhancing adsorption capacity while maintaining specific recognition performance of mesoporous silica: a novel imprinting strategy with amphiphilic ionic liquid as surfactant. Nanotechnology 29:375604. https://doi.org/10.1088/1361-6528/aace10
Engler AC, Wiradharma N, Ong ZY, Coady DJ, Hedrick JL, Yang YY (2012) Emerging trends in macromolecular antimicrobials to fight multi-drug-resistant infections. Nano Today 7:201–222
Park J, Kadasala NR, Abouelmagd SA, Castanares MA, Collins DS, Wei A, Yeo Y (2016) Polymer–iron oxide composite nanoparticles for EPR-independent drug delivery. Biomaterials 101:285–295
Zakerzadeh E, Alizadeh E, Kafil HS, Hassanzadeh AM, Salehi R, Mahkam M (2017) Novel antibacterial polymeric nanocomposite for smart co-delivery of anticancer drugs. Artif Cell Nanomed B 45:1509–1520
Luo ZM, Jin K, Pang Q, Shen S, Yan ZQ, Jiang T, Zhu XY, Yu L, Pang ZQ, Jiang XG (2017) On-demand drug releasing from dual targeting small nanoparticles triggered by high intensity focused ultrasound enhanced glioblastoma targeting therapy. ACS Appl Mater Inter 9:31612–31625
Tziveleka LA, Bilalis P, Chatzipavlidis A, Boukos N, Kordas G (2014) Development of multiple stimuli responsive magnetic polymer nanocontainers as efficient drug delivery systems. Macromol Biosci 14:131–141
Wen YF, Oh JK (2015) Intracellular delivery cellulose-based bionanogels with dual temperature/pH-response for cancer therapy. Colloid Surface B 133:246–253
Luo Z, Cai KY, Hu Y, Li JH, Ding XW, Zhang BL, Xu DW, Yang WH, Liu P (2012) Redox-responsive molecular nanoreservoirs for controlled intracellular anticancer drug delivery based on magnetic nanoparticles. Adv Mater 24:431–435
Meng ZQ, Wei F, Wang RH, Xia MG, Chen ZG, Wang HP, Zhu MF (2016) NIR-laser-switched in vivo smart nanocapsules for synergic photothermal and chemotherapy of tumors. Adv Mater 28:245–253
Jalani G, Tam V, Vetrone F, Cerruti M (2018) Seeing, targeting and delivering with upconverting nanoparticles. J Am Chem Soc 140:10923–10931
Cao Y, Dong HF, Yang Z, Zhong XM, Chen Y, Dai WH, Zhang XJ (2017) Aptamer-conjugated graphene quantum dots/porphyrin derivative theranostic agent for intracellular cancer-related microRNA detection and fluorescence-guided photothermal/photodynamic synergetic therapy. Acs Appl Mater Inter 9:159–166
Chen H, Wang ZY, Zong SF, Chen P, Zhu D, Wu L, Cui YP (2015) A graphene quantum dot-based FRET system for nuclear-targeted and real-time monitoring of drug delivery. Nanoscale 7:15477–15486
Li JH, Fu JQ, Yang Q, Wang LY, Wang XY, Chen LX (2018) Thermosensitive molecularly imprinted core-shell CdTe quantum dots as a ratiometric fluorescence nanosensor for phycocyanin recognition and detection in seawater. Analyst 143:3570–3578
Yu JL, Wang XY, Kang Q, Li JH, Shen DZ, Chen LX (2017) One-pot synthesis of a quantum dot-based molecular imprinting nanosensor for highly selective and sensitive fluorescence detection of 4-nitrophenol in environmental waters. Environ Sci-Nano 4:493–502
Wang XY, Yu JL, Li JH, Kang Q, Shen DZ, Chen LX (2018) Quantum dots based imprinting fluorescent nanosensor for the selective and sensitive detection of phycocyanin: a general imprinting strategy toward proteins. Sensor Actuat B-Chem 255:268–274
Amin M, Mahbub S, Hidayathulla S, Alam MM, Hoque MA, Rub MA (2018) An estimation of the effect of mono/poly-hydroxy organic compounds on the interaction of tetradecyltrimethylammonium bromide with levofloxacin hemihydrate antibiotic drug. J Mol Liq 269:417–425
Fuchs AV, Will GD (2010) Photo-initiated miniemulsion polymerization as a route to the synthesis of gold nanoparticle encapsulated latexes. Polymer 51:2119–2124
Li ZF, Wang ZH, Du XY, Shi C, Cui XJ (2018) Sonochemistry-assembled stimuli-responsive polymer microcapsules for drug delivery. Adv Healthc Mater 7:1701326. https://doi.org/10.1002/adhm.201701326
Du CB, Hu XL, Guan P, Gao XM, Song RY, Li J, Qian LW, Zhang N, Guo LX (2016) Preparation of surface-imprinted microspheres effectively controlled by orientated template immobilization using highly cross-linked raspberry-like microspheres for the selective recognition of an immunostimulating peptide. J Mater Chem B 4:1510–1519
Asnachinda E, Khaodhiar S, Sabatini DA (2009) Effect of ionic head group on admicelle formation by polymerizable surfactants. J Surfact Deterg 12:379–386
Alphandéry E, Idbaih A, Adam C, Delattre JY, Schmitt C, Guyot F, Chebbi I (2017) Development of non-pyrogenic magnetosome minerals coated with poly-l-lysine leading to full disappearance of intracranial U87-Luc glioblastoma in 100% of treated mice using magnetic hyperthermia. Biomaterials 141:210–222
Li YM, Lin JT, Zhi XL, Li PF, Jiang XF, Yuan J (2018) Triple stimuli-responsive keratin nanoparticles as carriers for drug and potential nitric oxide release. Mater Sci Eng, C 91:606–614
Wang MZ, Zhou CC, Chen J, Xiao YF, Du JZ (2015) Multifunctional biocompatible and biodegradable folic acid conjugatedpoly(epsilon-caprolactone)-polypeptide copolymer vesicles with excellent antibacterial activities. Bioconjugate Chem 26:725–734
Zhou CC, Wang MZ, Zou KD, Chen J, Zhu YQ, Du JZ (2013) Antibacterial polypeptide-grafted chitosan-based nanocapsules as an “Armed” carrier of anticancer and antiepileptic drugs. ACS Macro Lett 2:1021–1025
Ghanem OB, Mutalib MI, Elharbawi ME, Gonfa G, Kait CF, Alitheen NBM, Leveque JM (2015) Effect of imidazolium-based ionic liquids on bacterial growth inhibition investigated via experimental and QSAR modelling studies. J Hazard Mater 297:198–206
Aguirreurreta Z, CalL JCDL, Leiza JR (2017) Preparation of high solids content waterborne acrylic coatings using polymerizable surfactants to improve water sensitivity. Prog Org Coat 112:200–209
Koziróg A, Kręgiel D, Brycki B (2017) Action of monomeric/gemini surfactants on free cells and biofilm of asaia lannensis. Molecules 22:2036. https://doi.org/10.3390/molecules22112036
El Badawy AM, Silva RG, Morris B, Scheckel KG, Suidan MT, Tolaymat TM (2011) Surface charge-dependent toxicity of silver nanoparticles. Environ Sci Technol 45(1):283–287
Abbaszadegan A, Ghahramani Y, Gholami A, Hemmateenejad B, Dorostkar S, Nabavizadeh M, Sharghi H (2015) The effect of charge at the surface of silver nanoparticles on antimicrobial activity against gram-positive and gram-negative bacteria: a preliminary study. J Nanomater 2015:720654. https://doi.org/10.1155/2015/720654
Wu W, Yao W, Wang X, Xie C, Zhang JL, Jiang XQ (2015) Bioreducible heparin-based nanogel drug delivery system. Biomaterials 39:260–268
Xin C, Yao XZ, Du B, Yang WY, Wang LG, Ma LR, Weng WY (2018) Stearic acid-grafted chitooligosaccharide nanomicelle system with biocleavable gadolinium chelates as a multifunctional agent for tumor imaging and drug delivery. Pharm Res 36:10. https://doi.org/10.1007/s11095-018-2530-2
Alibolandi M, Ramezani M, Sadeghi F, Abnous K, Hadizadeh F (2015) Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int J Pharm 479:241–251
Li HJ, Zhu JX, Chen S, Jia L, Ma YL (2017) Fabrication of aqueous-based dual drug loaded silk fibroin electrospun nanofibers embedded with curcumin-loaded RSF nanospheres for drugs controlled release. Rsc Advances 7:56550–56558
Shi YF, Lv HL, Lu XF, Huang YX, Zhang Y, Xue W (2012) Uniform molecularly imprinted poly(methacrylic acid) nanospheres prepared by precipitation polymerization: the control of particle features suitable for sustained release of gatifloxacin. J Mater Chem 22:3889–3898
Marcelo G, Ferreira IC, Viveiros R, Casimiro T (2018) Development of itaconic acid-based molecular imprinted polymers using supercritical fluid technology for pH-triggered drug delivery. Int J Pharm 542:125–131
Luo YA, Cai XL, Hi L, Lin YH, Du D (2016) Hyaluronic acid-modified multifunctional Q-Graphene for targeted killing of drug-resistant lung cancer cells. Acs Appl Mater Inter 8:4048–4055
Du C, Hu X, Zhang G, Cheng Y (2019) 2D materials meet biomacromolecules: opportunities and challenges. Acta Phys -Chim Sin 35:0001–0009
Rong Q, Zhang YM, Wang C, Zhu ZQ, Zhang J, Liu QJ (2017) A high selective methanol gas sensor based on molecular imprinted Ag-LaFeO3 fibers. Sci Rep-UK 7:12110. https://doi.org/10.1038/s41598-017-12337-z
Masoumi M, Jahanshahi M (2016) Synthesis and recognition of nano pore molecularly imprinted polymers of thymol on the surface of modified silica nanoparticles. Adv Poly Tech 35:221–227
López TDF, García MED, Laíño RB (2014) Molecularly imprinted silica-silver nanowires for tryptophan recognition. Nanotechnology 25:425705. https://doi.org/10.1088/0957-4484/25/42/425705
Kim H, Lee D, Kim J, Kim TI, Kim WJ (2013) Photothermally triggered cytosolic drug delivery via endosome disruption using a functionalized reduced graphene oxide. ACS Nano 7:6735–6746
Mazurek P, Brook MA, Skov AL (2018) Glycerol-silicone elastomers as active matrices with controllable release profiles. Langmuir 34:11559–11566
Wen N, Dong YS, Song R, Zhang WP, Sun C, Zhuang XM, Guan Y, Meng QB, Zhang YJ (2018) Zero-order release of gossypol improves its antifertility effect and reduces its side effects simultaneously. Biomacromol 19:1918–1925
Acknowledgements
The authors are grateful for the financial support provided by the National Natural Science Foundation of China (Grant No. 51433008), and the Seed Foundation of Innovation and Creation for Graduate Students in Northwestern Polytechnical University (Grant No. ZZ2018185). In addition, the authors are grateful for the technical support provided by Analytical & Testing Center of Northwestern Polytechnical University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, Y., Hu, X., Guan, P. et al. A novel controllable molecularly imprinted drug delivery system based on the photothermal effect of graphene oxide quantum dots. J Mater Sci 54, 9124–9139 (2019). https://doi.org/10.1007/s10853-019-03500-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03500-0