
Abstract
Electrospun nanofibrous membranes (ENMs) are used in a variety of applications, including sensors, tissue engineering, air filtration, energy, and reinforcement in composite materials. Recently, they have gained an interest in the field of liquid filtration. The membranes, surface, bulk, and overall architecture play an important role in the filtration properties and hence the right characterization technique needs to be established, which will pave the way for future developments in the field of filtration. In this article, we have reviewed the recent advances in ENMs for liquid separation application.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
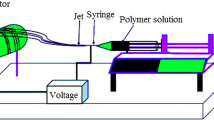
Electrospinning has been exploited for almost a century to process polymers and related materials into nanofibers with controllable compositions, diameters, porosities, and porous structures, for a variety of applications. Electrospinning is a relatively non-material specific process of fabricating nanofibers and its principle is well understood. A high voltage is required to elongate the polymer solution/melt from the tip of the spinneret and fibers can be collected in different assemblies depending on the type of collector [1]. Owing to its high porosity and large surface area, a nonwoven mat of electrospun nanofibers is highly explored in several applications such as tissue engineering and has been commercially used as air filters and recently it has gained interest in the area of liquid filtration.
An advantage of electrospun nanofibrous membrane (ENM) is that a variety of modification techniques can be applied on it to meet a certain desirable function. The nanofiber itself can also be functionalized through attachment of a bioactive species for affinity separation [2], and the top most surface can be modified by plasma-induced grafting [3], coating [4], or interfacial polymerization [5]. Different fiber diameters can be achieved by manipulating the processing parameters [1]. In addition, the nanofibers can be assembled into a variety of arrays or architectures by manipulating their alignment, stacking, or folding. All these attractive attributes make electrospinning a powerful tool for generating nanostructured material. Hence, researchers with various backgrounds such as science, engineering, and mathematics together with industry researchers, are working on this electrospinning area, which has resulted in significant advancements in this field. Figure 1 depicts the rapid growth of nanofibers since 2000 in terms of published articles (source SciFinder). An overview of nanofibers applied in various subjects and their application domain is given in Fig. 2. The application of ENM in water filtration domain is rapidly growing.

Number of journal and patent publications

Overview of the number of publications of nanofibers in various applications
Depending on the pore size of the materials, ENM have been applied in microfiltration (MF) [2, 3, 6], ultrafiltration (UF) [4, 7, 8], nanofiltration (NF) [5, 9, 10], and forward osmosis (FO) [11] applications, except reverse osmosis (RO). ENMs when applied as such showed enhanced flux than conventional membranes in MF applications. The flux was further improved by blending [6], surface modification [6], and in situ polymerization [12] techniques. The thin-film coating of MF membranes by chitosan were successfully applied in UF applications [7]. Several authors have reported that when the mid-layer made up of electro-spun MF/UF membranes was coated with top layer by thin-film composite configuration in NF; higher permeate flux for ENM composite than conventional membranes were achieved [5, 9, 10]. Optimization of various properties such as surface architecture, thickness, and pore size of the mid-layer ENM was studied to increase the flux and salt separation. Yoon et al. reported that TFNC membranes fabricated with ENM showed 2.4 times more permeate flux than conventional membranes in NF application, which is due to large open pore structure and low hydraulic resistance of ENMs [5]. The improvement in flux and separation efficiency by pore size reduction was reported by Kaur et al. [13]. The internal concentration polarization bottleneck observed in commercial phase-inverted-forward osmosis (PI-FO) membranes for desalination application was eliminated by applying ENM as support layer by Song et al. [11], which provides low tortuosity and high porosity.
Many reviews [14–31] have been written to give a snapshot of the recent developments of ENM in liquid filtration. Although these reviews address overall advancements in this field, a review article emphasizing the characterization of ENMs is very much needed as the surface and morphological properties of ENMs are quite different from the bulk materials. Also, some of the characterization involves significantly different sample preparation conditions when compared to bulk sample preparation (for example, tensile strength analysis). In addition, accurate characterization would be highly useful to develop the membranes with desired properties. Based on the desired properties, desired applications can be decided or fine-tuned. Hence, in this review, we have taken the opportunity to write about the various characterization techniques used to determine the properties of ENMs. When the ENMs are explored for liquid filter applications, several challenges faced and various electrospinning parameters and membrane modifications applied for its successful application are highlighted.
We have gathered a snapshot of various characterization techniques that have been established within our research group and other groups. These characterization techniques have proved to be useful and essential in determining the application of ENMs for liquid separation.
Membrane properties
Some of the interesting features of ENM are (1) high porosity due to interconnected structure and hence high flux, (2) enhanced specific surface area hence better efficiency, (3) unique topography hence unique features such as enhanced hydrophobicity, and (4) differing surface chemical properties and chemical composition. The above properties can be altered by changing the variable in the electrospinning process. The selection of the polymers, and choice of the solvent, this also affects the wettability/hydrophilicity of the membranes, which in turn decides the filtration properties of the ENM.
Two key parameters—flux and selectivity—define membrane efficiency in water filtration applications. Selectivity is governed by the surface properties of the membrane that discriminates the type of species that can pass through the membrane. Flux, on the other hand, describes how much gets into the membrane and how fast it moves. These two parameters, however, are affected by a number of factors and it is detrimental to characterize the membrane in relation to these.
Membrane characterization is useful to the determination of structural and morphological properties of the membrane. This includes knowledge of porosity, largest pore size (bubble point), pore-size distribution, wettability (hydro-phobicity/-philicity), transmembrane pressure drop across the membrane, membrane thickness, cross-sectional geometry, and flux permeability. The information gathered is essential in determining which type of separation processes the membrane can be used for and importantly its potential performance. Before the required properties are determined, the right characterization techniques need to be established and usually several techniques are required in conjunction with each other to determine the properties of the membrane.
Atomic force microscopy, differential scanning calorimeter, Fourier transform infrared spectroscopy, scanning electron microscopy, tensile test, transmission electron microscopy, and X-ray photoelectron spectroscopy
Atomic force microscopy (AFM) and scanning electron microscopy (SEM) observations directly give visual information on the membrane morphology, such as surface pore size and its distribution, pore density, surface porosity, and cross-sectional structure. Differential scanning calorimetry (DSC) is used to determine the thermal properties of ENMs and its rigidity. ENMs are rigid if the glass transition temperatures are high. The tensile strength of ENMs determines mechanical stability. Fourier transform infrared (FT-IR), and X-ray photoelectron spectroscopy (XPS) are used to find out the functional groups and elemental compositions of the ENMs. For example, the presence of functional groups, like amino (–NH) and hydroxyl (–OH), in the phenylcarbomylated β-cyclodextrin (CD) blended PMMA ENMs is confirmed by the FT-IR spectroscopy [32]. The existence of a nitrogen peak on the surface of ENMs confirmed the presence of phenylcarbomylated β-CD on the PMMA nanofiber surface, which was observed by XPS. Polyacrylonitrile (PAN) blended with multi-walled carbon nanotubes (MWCNTs) was used for ENMs [33]. It is observed from SEM images that after the incorporation of MWCNTs in the ENMs, the nanofibers are thinner and appear wavier as compared to the control PAN ENMs.
Heat treatment
When we started using electrospun membranes, it was very difficult to handle the membrane. This weak property became a stumbling block for the development of electrospun fibers as liquid filters. Specifically polysulfone (PSU) and polyvinylidene fluoride (PVDF) ENMs had a very loose “cotton-like” morphology and were not easy to handle. There were excessive surface charges on the fibers and hence they were easily detached from the layers below. To increase the integrity and strength, heat treatment was performed on both PSU and PVDF ENMs.
15 % (w/v) PVDF (MW 440 kDa, dissolved in 2:3 N,N′-dimethylacetylamide: acetone) was electrospun. The melting temperature of PVDF raw material and ENM was required so that the heat treatment temperature of the ENM could be determined. Thermal analysis of the PVDF raw material and ENM was performed on a DSC (Perkin Elmer, Pyris 6) in a temperature range of 20–200 °C at a rate of 10 °C/min. Table 1 shows the onset temperature, peak temperature of PVDF pellet and non-heat-treated PVDF ENM. The onset temperature was determined as 158 °C. Hence the heat treatment of the ENM (performed in a convection oven—carbolite) was applied before this temperature. When the membrane was heated directly to 157 °C, occasional fusion of several fibers occurred due to the low boiling point of acetone, which resulted in faster evaporation thereby inducing fusion to occur. This phenomenon is observed through SEM (Quanta 200F, FEI Comp., Hillsboro, OR, USA) in Fig. 3c. To prevent this, ENMs were heated from room temperature to 60 °C for 1 h at a rate of 1 °C/min to remove any trace amount of solvent. Subsequently, the membranes were then heated up to 157 °C at the same rate for 3 h to improve the structural integrity of the membrane. From the SEM micrographs in Fig. 3b it was evident that only overlapping fibers fused over one another. The heat-treated ENM was sufficiently flexible to be folded without any distortion to the membrane and is easily stamped out into a circular shape for water flux characterization at a later stage. Alternatively, the ENM could be heat treated at 60 °C for 1 h to remove any trapped residual solvent, and the temperature was subsequently raised to 150 °C at rate of 2 °C/min and maintained for 3 h.

SEM surface morphology of PVDF EM a before heat treatment, b after slow heat treatment, and c quick heat treatment
To further understand what happens to the structure of the heat-treated ENM, the heat-treated ENM (at 150 °C for 3 h) was subjected to thermal analysis. Figure 4 shows that the heat-treated ENM possessed two melting peaks unlike the native ENM and PVDF raw material. A double endotherm in the melting region on a DSC scan is generally found in semi-crystalline polymers. Two interpretations have been presented for this phenomenon. One is that the lower-temperature endotherm does not correspond to melting of a crystalline phase, but rather to a solid–solid phase transition or partial melting, and that the higher temperature endotherm indicates melting of the crystalline phase formed by such transitions as orientation changes of crystals, phase transition between crystalline modifications or recrystallization. The other interpretation is that the two endotherms were attributable to the melting of two different crystalline phases initially coexisting. Generally, solid phase PVDF can exhibit three different spherulitic crystalline forms: phase I, phase II, and phase III, depending upon the method of production [34]. The degree of crystallinity (χ) of PVDF normally ranges from 35 to 65 % [35, 36]. The χ does not go beyond 65 % due to two factors: the polymer is inherently easy to crystallize because of the simple structure of its monomer, but more complete crystallization is inhibited because of the presence of head to head structure in a polymer chain. The degree of crystallinity, χ, was calculated by the expression:
where H f is the heat of fusion for the tested sample and H 0f = 104.7 J/g [16], the heat of fusion for the 100 % crystalline sample. Table 2 reflects the χ of PVDF raw material, non-heat-treated PVDF ENM, and heat-treated PVDF ENM. It is clear that PVDF in raw material form has a low χ of 32 % and when it was electrospun into fibers, the χ increased by 17 %. However, when the ENM was heat treated, the χ increased by almost twice. This highlights the presence of more ordered structures within the fibers and the degree of orderness is increased further when the overall membrane was heat treated.

Melting transition of PVDF raw material, non-heat-treated ENM, and heat-treated ENM
The mechanical properties of the heat-treated and non-heat-treated ENM were tested in a tensile machine Instron 3345 equipped with a 100 N load cell. Three strips of length 3 cm by 1 cm were used for the tensile test.
The heat-treated membrane became sufficiently rigid to be used for further characterizations. The increase in rigidity is evident from the tensile profile shown in Fig. 5. The heat-treated PVDF membrane had a significantly higher mechanical strength (8.5 MPa) compared to the non-heat-treated PVDF ENM (0.4 MPa).

Mechanical strength of heat-treated and non-heat-treated ENM [3]
Likewise the melting temperature (T m) of PSU ENMs was measured using DSC. Based on DSC measurements, the T g (glass transition temperature) and T m of PSU (Mn 26,000 Da, Aldrich, St Louis, MO, USA) was determined to be identical, occurring at 190 °C (Fig. 6a). This is quite unique. Based on this information, heat treatment of 20 % (w/v, dissolved in N,N′-dimethylformamide) PSU ENM was conducted just below 190 °C (i.e. at 188 °C) for 3 h. This caused the fibers to “fuse” together and thereby intersect each other and hence increase the structural integrity of the overall membrane. This is supported by tensile test study using Instron 3345 as shown in Fig. 6b where the mechanical strength of the heat-treated membrane was increased by threefold. Upon heat treatment, the integrity of the membrane was significantly improved. It was easily handled; changing from a cotton-like morphology to that of paper. The observed SEM images (Fig. 6c, d) confirm this concept, which was further supported by AFM images (Fig. 6e). Thermal analysis of the non-heat-treated and heat-treated PSU membranes were performed on a DSC. As evinced from Fig. 6a heat-treated PSU showed an increased crystallinity as compared to non-heat-treated PSU. Having successfully improved the handling of the fibrous mat, it was now possible to treat it as a separating unit—a membrane filter—and study its properties.

a Thermal analysis of PSU pellet and PSU heat-treated ENM, b mechanical strength of heat-treated PSU ENM and untreated PSU nanofiber, c SEM micrograph of non-heat-treated PSU ENM, d SEM micrograph of heat-treated PSU ENM, e AFM of heat-treated PSU ENM
Hot pressing
One of our studies has shown that the hot pressing of PAN ENM [9] prior to interfacial polymerization is essential as it “locks” the fibers in place and improves the adhesion of the ENM layer with the backing material (BM). The benefit of this treatment was twofold. It was realized through SEM micrographs (Fig. 7) that the adhesion between the ENM layer and BM improved. When the membrane was not hot pressed, a “loose” network of fiber structure was observed. Second, the thickness of the ENM layer becomes more compact. Mechanical properties of these membranes were studied and it was revealed that as the pressure of the hot pressing machine increases, the thickness of the ENM layer reduces and the tensile yield improves.

Cross-sectional images of a non-hot-pressed PAN ENM and b hot-pressed PAN ENM. Dotted arrow indicates the “loose” and non-compact structure of the ENM layer. Full red arrow indicates the improved adhesion between the ENM layer and backing material [9] (Color figure online)
Subsequently when a thin film is formed on the surface of the hot-pressed ENM via interfacial polymerization (labeled as TFNC, thin-film nano-composite), the rejection of 2000 ppm magnesium sulfate is considerably high and stable even at high temperatures as compared to non-hot-pressed ENM.
AFM results revealed that surface roughness values of hot-pressed TFNC were reduced as compared to non-hot-pressed TFNC.
Membrane thickness
The thickness of a membrane is one of the important factors that influence flux. The thickness of the selective layer (that governs the separation) is an integral component of transport models such as Pore Model [37], used for solute transport through pores or Thomas Model used for affinity separation [38]. Thus, independent of the type of separation that occurs, it is important to measure this feature as accurately as possible.
Although it would seem to be a simple feature to measure, this is far from being that the case. The most straight forward method would be to use a micrometer, if the membrane thicknesses are around 50–200 μm. However, when the membrane is highly porous, like that of a nanofibrous membrane, using a micrometer can lead to erroneous measurements due to compression of the membrane.
One of the methods that can measure the thickness of the ENM would be using a microscope. The advantage of this method is that the cross-sectional geometry of the membrane can also be observed simultaneously (in addition to thickness). We have explored few alternative techniques (on PSU ENM) which are presented below.
Cryotome and microtome sectioning
The first technique carried out was to embed the membrane in a mounting medium and then sectioning it on a cryotome. This is a common technique used in biological research to observe the cross-section of tissues. Very thin sections that are of several microns are possible to measure.
PSU ENM was covered with a tissue mounting medium, which is in a solution state at room temperature. This was then quenched into liquid nitrogen to freeze the sample. The sample was then transferred into a cryotome that was maintained at −20 °C and sliced into 10 μm sections. The sections were collected onto glass cover slips and upon removal from the cryotome, the solution melted and the membrane section recovered. The section was then gold coated and observed under SEM. A typical image is shown in Fig. 8.

Cryotome sectioned of PSU ENM a ×400 magnification and b ×3000 magnification
Although it was possible to measure the thickness of the membrane, the geometry of the pore spaces across the membrane is not visible and was masked. Thus, this method was not as effective as expected.
Another mounting medium, paraffin wax, was considered. This time the sectioning was done on a microtome as sectioning at room temperature. Unfortunately, this method was even less informative than the previous method. From Fig. 9, it can be seen that neither thickness nor geometry was visible. Since both these attempts failed, it was concluded that embedding the method was not a suitable method for ENMs.

Microtome sectioned PSU ENM
Quenching in liquid nitrogen
To section the membrane without any embedding medium, quenching it into liquid nitrogen was considered. The temperature of liquid nitrogen is −196 °C. It was expected that at such low temperatures, materials will become brittle and thus, can be easily fractured. However, the ENMs were still flexible.
Even after prolonged immersion, it did not become brittle. This was a very surprising observation. This was repeated many times with prolonged immersion, but the ENM still remained flexible.
Blade sectioning
As all the above methods proved to be unsuccessful, the membrane was then cut with an extremely sharp surgical blade. The resulting images are shown in Fig. 10.

Blade-sectioned of PSU ENM at a ×150 magnification, and b ×1200 magnification
The blade sectioning was by far the best method for measurement. The PSU membrane thickness from the SEM image (Fig. 10) was 240 μm. When the same membrane was measured using a micrometer, the thickness was 190 μm. As can be seen from the above measurements, the difference is very large. Such large errors in the measurement may lead to very poor model analysis.
Cross-section geometry
Another observation obtained from the above image (Fig. 10) is that the “flow” path across the ENM would be extremely tortuous; no clear interconnecting pore structures were visible. There was no “clear” path for solute to transport across the ENM. Most transport models developed for membrane processes rely on the assumption of a continuous pore across the membrane thickness. Although most of the membranes do not possess such a characteristic, independent of the fabrication technique used, the assumption might be more invalid for ENMs. If this proves to be so, a corrective parameter must be included or a new transport model has to be developed. Such information can only be derived with the experimental data generated through the course of many studies similar to this.
When the ENM was sectioned, the SEM image revealed several kinds of morphologies. Two kinds of fiber morphologies were observed. When the fiber diameter was smaller (less than 300 nm), it appeared smooth. However, as the fiber diameter increased, it appeared to take on a convoluted surface. There are two possible scenarios for this difference. The lack of a smooth surface could be due to the occurrence of phase separation taking place. When the fiber is very small, possibly much of the solvent had already “escaped” and there is no generation of solvent-rich or solvent-poor regions that can induce such features with time. In the larger fibers, there is more mass present and it is possible for phase separation to occur.
Closer inspection of some of the larger fibers indicates that these fibers seem to be formed from the fusion of several smaller fibers, which could have occurred during the heat treatment process. This could explain the presence of some larger fibers having the convoluted fibers. It can be observed from the SEM image (Fig. 11) that both scenarios exist. Some large fibers have convoluted surfaces that seem to be ordered along the length of the fiber, indicating that it was formed due to the fusion of smaller fibers. Simultaneously there are large fibers that have a highly convoluted surface which indicates the probability of phase separation, similar to those of beads.

Morphologies observed: a fiber diameter and b bead showing porous ultrastructure [1]
Even more interestingly, the bead exhibited a unique ultrastructure within (Fig. 11b). The SEM image suggests the possibility of some form of phase separation that is taking place within the bead (Fig. 11b).
The above observations indicate that the humidity level and the choice of the solvents could be two of the important parameters that affect the spinning for smooth uniform fibers. On the other hand, when an increased surface area is preferred, it might be better to spin at high humidity levels to attain such convoluted morphology.
Hydrophobic/hydrophilic characteristics
The hydrophobic (or hydrophilic) nature of a membrane has a direct impact on the avenue of its usage. For example, in the case of a membrane distillation domain, it is desirable to use a hydrophobic membrane, as the water must not enter the membrane. On the other hand, in the case of separation of protein solutions, hydrophilic membranes are preferred, if possible, to minimize protein adsorption. Hence after the fabrication of ENMs, they were subjected to water contact angle (WCA) measurements. The WCA is the measure of the angle formed between the surface of a solid and the line tangent to the droplet radius from the point of contact with the solid. The WCA is related to the surface tension by Young’s equation through which the behavior of specific liquid–solid interactions can be calculated. In general, the contact angles are measured by various techniques, such as the static and dynamic sessile drop method, dynamic Wilhelmy method, etc.
A WCA of zero results in wetting, while an angle between 0° and 90° results in spreading of the drop (due to molecular attraction). Angles greater than 90° indicate the liquid tends to bead or shrink away from the solid surface, i.e., surface is hydrophobic (Fig. 12).

Contact angle measurements: wetting of a solid surface
Advancing contact angle
VCA Optima Surface Analysis System (AST Products, Inc., Billerica, MA, USA) equipment was used to measure the WCA of the PSU membranes. The static advancing WCA measured was around 140°. This is almost 100 % higher than the WCA (60°–70°) reported for the same material in film form. The ENMs were exhibiting higher hydrophobicity characteristics than the conventional phase-inversion membranes. When a more hydrophobic material, such as PVDF, was electrospun, the resulting WCA was also observed to be around 145° (Fig. 13) [39] when compared to WCA of 80°–90° in film form.

Contact angles of electrospun a PSU and b PVDF ENM
The above results indicated that the morphology has significantly influenced the wetting behavior. In surface technology, two models exist that relate the surface morphology to hydrophobicity. These are the “Cassie Model” [40] and the “Wenzel Model” [41], named after the scientists who discovered and developed the models, respectively.
The Wenzel model relates the increased surface area, brought about by surface roughness, as the cause of an increased hydrophobicity in materials. The Cassie model on the other hand states that the air pockets in the hollows of a rough surface are the cause of the increased hydrophobicity. Both models, to a certain extent, are related and they are generally accepted arguments when justifying the hydrophobic phenomenon (Fig. 14).

Schematic of the Cassie and Wenzel model effects
The AFM and SEM images of ENM show that both attributes are present. There are large air pockets on the surface (Fig. 15a) and the roughness is in the micron range (Fig. 15b). These could have contributed to the increased hydrophobicity.

Attributes of PSU nanofibrous membrane that could contribute to increased hydrophobicity. a SEM micrograph. b AFM image and properties
The liquid entry pressure of water (LEPw) of the ENM was experimentally determined to be 1.80 psi (Fig. 16) [10]. Thus, there is initial “resistance” for water to enter a completely dry ENM. Once the ENM is completely wetted, the high porosity contributes to relatively large flow rates and the pressure can be reduced.

Flux–pressure profile of PSU-ENM [10]
More in-depth experimentation is required to determine which of these parameters have a dominant effect on the hydrophobicity of the membrane. Different materials—hydrophilic and hydrophobic—must be considered to see if such a phenomenon exists or if it is just pertaining to hydrophobic materials. Once the dominant parameter is identified, ENM can find applications in membrane distillation, where hydrophobicity and high porosity are essential.
Hydrophilicity can be improved by surface modification techniques such as plasma-induced grafting [3]. The surface of PVDF-ENM was exposed to argon plasma and subsequently graft-copolymerized with methacrylic acid. The surface-modified membrane achieved a 0° WCA. In addition to an improved surface hydrophilicity, the surface pores can be reduced depending on the plasma-induced grafting parameters. PVDF was blended with polyurethane-based hydrophilic surface modifying macromolecules (LSMMs) to make ENMs and the in-depth investigation was made regarding characterization of ENMs [6]. LSMM of end-groups PEG of average molecular weight 1000 Da (SMM-1000) played a significant role in increasing the hydrophilicity of the PVDF blended ENM, when compared to LSMM of end-groups PEG of average molecular weight 400 and 600 Da (LSMM-400 and LSMM-600) of slightly increased hydrophobicity. This phenomenon was due to the orientation that the LSMMs adopted during electro-spinning in which the hydrophobic part of LSMM-400 and LSMM-600 was primarily at the surface as compared to LSMM-1000. Recently, the modified PVDF hydrophobic composite membranes were made using hydrophobic surface modifying macromolecules (BSMM) through electrospun process and tested for direct contact membrane distillation (DCMD) with feed 3.5 wt% NaCl solution [42]. Electrospun-modified PVDF membranes have higher WCAs than the control PVDF membrane. The pore size of the nano-fiber membrane increases with BSMM concentration. The average fiber diameter of the modified PVDF-BSMM nano-fiber membranes increased with an increase in BSMM concentration. Acrylic acid and methacrylic acid were grafted to PVDF ENMs by plasma-induced or chemically induced copolymerization or a combination of both [43]. The presence of grafting on the top surface of the ENMs was confirmed by the WCA and XPS observations.
Electrospun nanofiber membranes consisting of PVDF blended with clay were prepared to prevent the pore wetting by adding highly hydrophobic clay into the dope solution and characterize the PVDF–clay nanocomposite electrospun membranes with different clay concentrations were characterized and also tested for DCMD [44]. It was noticed that the WCA of the membranes increased as the concentration of clay increased and at the highest clay concentration of 8 wt% a membrane with WCA of 154.2 ± 3.0° was obtained. It is noted that the melting point of the PVDF–clay electrospun nanofiber membrane increases with the increasing concentration of clay which indicated that the clay particles influence the crystallization process of the nanocomposite membrane.
PVDF nanofiber membrane is used in air-gap membrane distillation to produce potable water from saline water and also removal of chloroform from water was carried out by membrane gas stripping [45, 46]. The WCA of the PVDF nanofiber membrane was 130° and the AFM images were obtained wherein the surfaces were graded due to the presence of nodules. It is noted that these nodules are aligned and elongated toward the axial direction of nanofiber [45].
Transmembrane pressure
The flow rate across a membrane depends on the transmembrane pressure differential. Transmembrane pressure is the pressure difference across the top and bottom sides of the membrane. The lower the pressure difference, the easier is the flow through the membrane (less resistance to flow). To determine the flux through the nanofibrous membranes, a simple set-up was constructed, the schematic of which is shown in Fig. 17a.

Schematic of a permeation cell and b ENM configuration placed within the cell
One of the early challenges faced was to find an effective way to place the ENM into a permeation cell. When the ENM was placed directly into the cell, the edges of the cell were cutting the ENM. To protect the ENM, an aluminum sheet with a 25 mm diameter hole was glued to the ENM (Fig. 17b). This proved to be effective. In the first attempt, a stainless steel module of 200 ml capacity was used. The membrane was placed as shown in the schematic and nitrogen gas was used to pressurize the system until water started to flow out at the outlet.
It was observed that very little pressure was needed for the PSU-ENM used. The pressure was much less than 1 bar. The set-up was not able to measure this small drop. The air flow could not be regulated effectively at lower pressures. To achieve more control and allow for small incremental increases of pressure, the set-up was modified to include a reservoir gas tank, as shown in Fig. 18. This set-up was successful in determining the flow rate effectively with sufficient control. All flux studies were carried out using this set-up.

Schematic of a modified permeation cell set-up suited for ENMs [37]
From the affinity membrane point of view, having such a low pressure drop is advantageous. This eliminates the need for pressurizing the system during separation. However, since the pressure drop is directly proportional to the membrane thickness, there is a high chance that this will increase if several ENM sheets are used for the separation. Thus it is important to study the pressure drop as a function of membrane thickness.
Pore size, pore-size distribution, and porosity
Pore size refers to the diameter of the pores present in the membrane. Such information can discriminate between the type (size or molecular weight) of species which can permeate through and will be retained. However, pores in the membrane, especially polymeric membrane, do not have identical pore size but rather a range of sizes. This is known as the pore-size distribution. Whereas, porosity is the fraction of the membrane volume occupied by the pores (void volume). The pore size and its distribution discriminates the type of species that can permeate and it is the porosity that determines the flux.
Liquid intrusion technique—mercury porosimeter
The most common method employed in membrane technology to determine pore size and its distribution is the Mercury Intrusion method. Mercury has a high surface tension (485.5 dynes/cm at 2 °C) [47] and forms large contact angles with most other materials (∼=130°) [48] and is therefore considered a non-wetting liquid. Mercury does not spontaneously penetrate pores by capillary action so an external pressure must be applied to force the mercury into the pores. Mercury intrusion measures the pressure required to push mercury through the pores in a material. At low pressures, the larger pores get filled first. This transition pressure can be related to pore size by the Washburn equation described in the well-known Ref. [49]. As the pressure increases, the smaller pores get filled. Through this relationship the pore size and its distribution can also be obtained. Furthermore, by measuring the volume of mercury pushed into the membrane, the porosity can be determined. The high pressure required to force viscous mercury through the pores distorted the ENM. A similar finding has been reported that mercury intrusion causes an irreversible structural compression [50]. The mercury intrusion method can only provide useful representations of porosity and surface area [47, 51]. In addition the danger of using mercury makes the method unattractive.
Liquid extrusion technique: capillary flow porometer
In the case of ENMs, a more suitable approach to obtain the pore size and distribution would be to use a liquid extrusion technique instead of intrusion. In this approach, a wetting liquid is used to fill all the pores of the sample. The wetting liquid should possess a liquid/solid surface free energy (γl/s) that is less than the solid/gas surface free energy (γs/g). Therefore filling of the pores is spontaneous, but removal of the liquid from the pores is not. Pressure (from a non-reacting gas) is applied on the sample to remove the wetting liquid from the pores and permit gas flow.
Capillary flow porometry is based on this technique. In a capillary flow porometry measurement, a non-reacting gas (typically air) flows through a dry sample and then through the same sample after it has been wet with a liquid of known surface tension. The change in flow rate is measured as a function of pressure for both dry and wet processes. Because of the low pressure applied during the process, the porous structure of nanofiber membranes is not distorted [52]. Thus, capillary flow porometry can provide reproducible pore size and its distribution measurements with ignorable distortion errors. All the required pore structure characteristics such as pore size at bubble point, mean flow pore, and pore-size distribution can be computed from the measured differential pressures and gas flow rates. In this study, a capillary flow porometer (Porous Materials Inc., Ithaca, NY, USA) and a wetting liquid, Galwick™ was used.
The relationship between the pore size and the corresponding pressure is given by the Young–Laplace Eq. (2):
where R is radius of the pore, ΔP is differential gas pressure, γ is surface tension of wetting liquid, Galwick™ (γ = 15.9 dynes/cm) and θ is wetting angle.
The pressure at which the capillary action of the fluid within the largest pore is overcome is termed as the bubble point pressure. This bubble point pressure is used to determine the largest pore the ENM possesses using Eq. (2). The mean flow pore diameter is computed from mean flow pressure. The mean flow pore diameter is such that 50 % of flow is through pores larger than the mean flow pore diameter and 50 % of flow is through pores smaller than the mean flow pore diameter.
The wetting liquid, Galwick™, spontaneously wets the PSU-ENM (fiber diameter and thickness of PSU-ENM was 470 nm and 135 nm, respectively) and hence the wetting angle was set at θ = 0. Figure 19 shows the typical wet and dry curves obtained by the porometer run and Fig. 20 reflects the pore-size distribution of PSU-ENM. The wet and dry curves meet at higher pressures indicating that all the pores within the membranes have been completely “opened” during the porometer run.

Dry and wet curves of PSU-ENM obtained from “Capillary Flow Porometer” [10]

Pore-size distribution histogram of PSU-ENM used for study [10]
The run was repeated three times for each membrane to validate the accuracy of the measurement technique. For each membrane, the repeated runs produced similar bubble points and the smallest pore measurements. This highlights that ENMs have sufficient rigidity to undergo repeated testing cycles without undergoing severe compression or distortion.
Particle-challenge test
A particle-challenge test [10] was done to determine the separation characteristic of ENM. 100 ppm of polystyrene (PS) microparticles (Sigma-Aldrich, St. Louis, MO, USA) of 0.1, 0.5, 1, 2, 3, 7, 8, or 10 µm in size were used to identify the separation characteristics of PSU-ENM (with pore size of 4.6 µm) and to relate it to the pore-size distribution determined by capillary flow porometer.
The presence of any PS microparticles in permeate were detected via total organic carbon (TOC) Combustion Analyzer (Apollo 9000, Teledyne Tekmar, Mason, MO, USA) and the concentration was determined using the calibration curve obtained for each particle size. The separation experiment was stopped after 30 mL of permeate was collected. The remaining PS solution was removed and the cell was rinsed three times, without removing the PSU-ENM. The cell was refilled with distilled water and the water flux was determined again. Rejection (R %) was determined using the Eq. (3) below:
where C permeate and C feed are the PS solution concentration of permeate collected and the original feed, respectively. In addition, the flux recovery after each separation was also determined (Eq. 4). It is the ratio of the pure water flux after separation and before separation of a microparticle, X.
Separation was carried out first with the 10 µm followed by 8, 7, 3, 2, 1, 0.5 µm and finally 0.1 µm at 0.5 psig. The membrane was not changed throughout this experiment to understand the effectiveness as well as the reusability of the ENMs. The separation experiment was designed to give insight into the challenges. The effectiveness of the ENMs for removal of microparticles and the limiting particle size beyond which it fails to separate the microparticles was considered. Through the experimental findings, it was realized that when the microparticles are larger than the pores within the ENM, the membrane acts as screen filter and no fouling was observed. The ENM was most severely and irreversibly fouled when separating 2 and 1 µm, which corresponded to the mean flow pore diameter of the ENM. When sub-micron particles were separated, they tended to be attracted to the surface of the nanofibers and thus the ENM acts as a depth filter. The attraction of these particles onto the surface of the nanofibers can occur by numerous mechanisms: direct impaction, inertial impaction, and diffusion. Direct impaction occurs when a particle collides head-on with one of the fibers. Inertial impaction results if the particle in the fluid stream fails to negotiate the tortuous path presented by the random fiber in the membrane, which collides with and adheres to a fiber. Diffusion (Brownian motion) occurs when extremely small particles “flow through” the membrane regardless of fluid flow, where they are likely to be retained by adsorptive forces. Such Brownian movement is typical of particles less than 1 µm [53]. The above separation characterization highlights the potential of ENMs as barrier materials in separation technology. With a better understanding of their separation behavior, efficient pre-filters can be designed and developed, taking advantage of their large internal surface area. Such pre-filters should be used in various applications such as removal of microparticles from waste-water and with ultra-filtration or nano-filtration membranes, to prolong the life of these membranes.
The particle-challenge test also highlighted the robustness of the ENMs. Throughout the test, the membrane was structurally intact. More importantly, the separation results obtained and the properties of ENM determined through the characterization methods employed had a high correlation. This indicates that the characterization techniques employed proved to be an accurate means to predict the performance of an ENM with respect to size-exclusion separation processes. A similar study has also been performed on PVDF ENM [38].
Brunauer–Emmett–Teller (BET) gas adsorption
The surface area and pore structure of the materials determined through BET adsorption method is more reliable method than capillary flow and mercury porosimetry methods. This is due to the assumption of cylindrical pore geometry by the later methods. The BET surface area was found to be dependent on the average fiber diameter and smaller fiber diameter membranes have the large specific surface area. The total pore area range varied between 51 and 9 m2/g for average fiber diameter ranges of 167–2737 nm [51].
Transmission electron microscopy (TEM)
TEM has been widely used to characterize the incorporation of nanoparticles [54], MWCNTs [55] and core–shell nanostructures of electrospun membranes [54]. The incorporation of boehmite nanoparticle flake mounted on an electrospun nylon fiber was studied by Hota et al. [54] and applied for the removal of Cd(II) metal ions in water. The MWCNTs encapsulation enhanced the mechanical durability of polyacrylic acid/polyvinyl alcohol nanofiber and studied their Cu(II) removal ability [55].
X-ray energy dispersive spectroscopy (EDS)
EDS has been widely used to study the encapsulation of various nanostructures such as nanoparticles into the nanofibers matrix [55]. It has also been used to confirm the adsorption of various metal ions on to nanofiber surface from water solution.
X-ray diffraction (XRD) analysis
The crystalline or amorphous nature of the ENMs and ceramic nanofibrous membranes were evaluated by XRD analysis. Generally, the ENMs such as PSU and PVDF were found to be amorphous after electrospinning [56]. Miao et al. reported that electrospun ceramic nanofiber such as SiO2 nanofiber was amorphous. After decoration of this nanofiber by boehmite, core/sheath nanofibers (SiO2 as core and boehmite as shell) were formed in which peaks related to inorganic component was observed on the surface of SiO2 [57] and tested for water remediation. XRD technique has also been successfully applied to examine the incorporation of nanoparticles within nanofiber matrix by Hota et al. [54] and studied for the removal of metal ions.
Mass spectral analysis
Royen et al. applied static secondary ion mass spectrometry (S-SIMS) for the surface analysis of nanofibers [58]. Molecular composition at the surface of poly(ε-caprolactone) (PCL) nanofibres containing 0–15 mol % (relative to PCL) of cetyltrimethylammonium bromide (CTAB) was studied by S-SIMS spectra. They observed surface enrichment and depletion of the polar CTAB additive relative to the PCL matrix for low and high CTAB concentrations samples.
Conclusions
The first hurdle for developing an ENM was to maintain its structural integrity. This was successfully overcome by post-treatment of ENMs after electrospinning. Having overcome the handling issue, the next step was to find suitable characterization techniques. By making some modifications to traditional techniques and understanding the nature of ENMs, characterization techniques were established.
The advent of the capillary porometer was significant as it allows the pore-size distribution of ENMs to be effectively characterized. From the permeation studies, it was observed that some design changes in the permeation cell were needed to accommodate an ENM. However, without having to re-design, simple solutions such as adding aluminum ring over the ENM to protect it from the sharp edges proved to be effective. All the established techniques will allow for good characterization of developed ENMs and to understand the influence of architecture on a ENMs performance. Furthermore, along the way, interesting nuances of ENMs were discovered, which would play a significant role in determining the potential areas for application for ENMs in separation technology.
References
Ramakrishna S, Fujihara K, Teo WE, Lim TC, Ma Z (2005) An introduction to electrospinning and nanofibers. World Science, Singapore
Ma Z, Kotaki M, Ramakrishna S (2006) Surface modified nonwoven polysulphone (PSU) fiber mesh by electrospinning: a novel affinity membrane. J Membr Sci 272:179–187
Kaur S, Ma Z, Gopal R, Singh G, Ramakrishna S, Matsuura T (2007) Plasma-induced graft copolymerization of poly(methacrylic acid) on electrospun poly(vinylidene fluoride) nanofiber membrane. Langmuir 23:13085–13092
Wang X, Fang D, Yoon K, Hsiao BS, Chu B (2006) High performance ultrafiltration composite membranes based on poly(vinyl alcohol) hydrogel coating on crosslinked nanofibrous poly(vinyl alcohol) scaffold. J Membr Sci 278:261–268
Yoon K, Hsiao BS, Chu B (2009) High flux nanofiltration membranes based on interfacially polymerized polyamide barrier layer on polyacrylonitrile nanofibrous scaffolds. J Membr Sci 326:484–492
Kaur S, Rana D, Matsuura T, Sundarrajan S, Ramakrishna S (2012) Preparation and characterization of surface modified electrospun membranes for higher filtration flux. J Membr Sci 390–391:235–242
Yoon K, Kim K, Wang X, Fang D, Hsiao BS, Chu B (2006) High flux ultrafiltration membranes based on electrospun nanofibrous PAN scaffolds and chitosan coating. Polymer 47:2434–2441
Ma H, Yoon K, Rong L, Mao Y, Mo Z, Fang D, Hollander Z, Gaiteri J, Hsiao BS, Chu B (2010) High-flux thin-film nanofibrous composite ultrafiltration membranes containing cellulose barrier layer. J Mater Chem 20:4692–4704
Kaur S, Barhate R, Sundarrajan S, Matsuura T, Ramakrishna S (2011) Hot pressing of electrospun membrane composite and its influence on separation performance on thin film composite nanofiltration membrane. Desalination 279:201–209
Gopal R, Kaur S, Feng C, Chan C, Ramakrishna S, Tabe S, Matsuura T (2007) Electrospun nanofibrous polysulfone membranes as pre-filters: particulate removal. J Membr Sci 289:210–219
Song X, Liu Z, Sun DD (2011) Nano gives the answer: breaking the bottleneck of internal concentration polarization with a nanofiber composite forward osmosis membrane for a high water production rate. Adv Mater 23:3256–3260
Nabeela Nasreen SAA, Sundarrajan S, Syed Nizar SA, Balamurugan R, Ramakrishna S (2014) In-situ polymerization of PVDF-HEMA polymers, electrospun membranes with improved flux and antifouling properties for water filtration. Polym J 46:167–174
Kaur S, Sundarrajan S, Rana D, Matsuura T, Ramakrishna S (2012) Influence of electrospun fiber size on the separation efficiency of thin-film nanofiltration composite membrane. J Membr Sci 392:101–111
Barhate RS, Ramakrishna S (2007) Nanofibrous filtering media: filtration problems and solutions from tiny materials. J Membr Sci 296:1–8
Kaur S, Gopal R, Ng WJ, Ramakrishna S, Matsuura T (2008) Next generation fibrous media for water treatment. MRS Bull 33:21–26
Thavasi V, Singh G, Ramakrishna S (2008) Electrospun nanofibers in energy and environmental applications. Energy Environ Sci 1:205–221
Yoon K, Hsiao BS, Chu B (2008) Functional nanofibers for environmental applications. J Mater Chem 18:5326–5334
Chu B, Hsiao BS (2009) The role of polymers in breakthrough technologies for water purification. J Polym Sci Polym Phys Ed 47:2431–2435
Ramakrishna S, Jose R, Archana PS, Nair AS, Balamurugan R, Venugopal J, Teo WE (2010) Science and engineering of electrospun nanofibers for advances in clean energy, water filtration, and regenerative medicine. J Mater Sci 45:6238–6312. doi:10.1007/s10853-010-4509-1
Yoshikawa M, Tanioka A, Matsumoto H (2011) Molecularly imprinted nanofiber membranes. Curr Opin Chem Eng 1:18–26
Balamurugan R, Sundarrajan S, Ramakrishna S (2011) Recent trends in nanofibrous membranes and their suitability for air and water filtrations. Membranes 1:232–248
Nataraj SK, Yang KS, Aminabhavi TM (2012) Polyacrylonitrile-based nanofibers—a state-of-the-art review. Prog Polym Sci 37:487–513
Sundarrajan S, Ramakrishna S (2013) New directions in nanofiltration applications—are nanofibers the right materials as membranes in desalination? Desalination 308:198–208
Schiffman JD, Schauer CL (2008) A review: electrospinning of biopolymer nanofibers and their applications. Polym Rev 48:317–352
Ma HY, Chu B, Hsiao BS (2012) Functional nanofibers for water purification. In: Wei Q (ed) Functional nanofibers and their applications. Woodhead, Cambridge, pp 331–370
Raghavan P, Lim D-H, Ahn J-H, Nah C, Sherrington DC, Ryu H-S, Ahn H-J (2012) Electrospun polymer nanofibers: the booming cutting edge technology. React Funct Polym 72:915–930
Wang X, Ding B, Sun G, Wang M, Yu J (2013) Electro-spinning/netting: a strategy for the fabrication of three-dimensional polymer nano-fiber/nets. Prog Mater Sci 58:1173–1243
Feng C, Khulbe KC, Matsuura T, Tabe S, Ismail AF (2013) Preparation and characterization of electro-spun nanofiber membranes and their possible applications in water treatment. Sep Purif Technol 102:118–135
Sundarrajan S, Balamurugan R, Kaur S, Ramakrishna S (2013) Potential of engineered electrospun nanofiber membranes for nanofiltration applications. Drying Technol 31:163–169
Hasanzadeh M, Hadavi Moghadam B (2013) Electrospun nanofibrous membranes as potential adsorbents for textile dye removal-A review. J Chem Health Risks 3:15–26
Zhang L, Aboagye A, Kelkar A, Lai C, Fong H (2014) A review: carbon nanofibers from electrospun polyacrylonitrile and their applications. J Mater Sci 49:463–480. doi:10.1007/s10853-013-7705-y
Kaur S, Kotaki M, Ma Z, Gopal R, Ramakrishna S, Ng SC (2006) Oligosaccharide functionalized nanofibrous membrane. Int J Nanosci 5:1–11
Singh G, Rana D, Matsuura T, Ramakrishna S, Narbaitz RM, Tabe S (2010) Removal of disinfection by-products in water by carbonized nanofiber membranes. Sep Purif Technol 74:202–212
Nakagawa K, Ishida Y (1973) Annealing effect in poly(vinylidene fluoride) as revealed by specific volume measurements, differential scanning calorimetry and electron microscopy. J Polym Sci Polym Phys Ed 11:2153–2173
Dohany JE, LE Robb (1980) Poly(vinylidene fluoride). In: Othmer DF (ed) Kirk-Othmer encyclopaedia of chemical technology, vol. 11, 3rd edn. Wiley, New York, pp 64–74
Glennon D, Smith JR, Nevell RT, Begg D, Mason SE, Watson KL, Tsibouklis J (1997) An atomic force microscopy study of the effect of tensile loading and elevated temperature on polyvinylidene fluoride from flexible oil pipelines. J Mater Sci 32:6227–6234. doi:10.1023/A:1018680909681
Matsuura T (1994) Synthetic membranes and membrane separation process. CRC, Boca Raton
Suen S-Y, Etzel MR (1992) A mathematical analysis of affinity membrane bioseparations. Chem Eng Sci 47:1355–1364
Gopal R, Kaur S, Ma Z, Chan C, Ramakrishna S, Matsuura T (2006) Electrospun nanofibrous filtration membrane. J Membr Sci 281:581–586
Cassie ABD, Baxter S (1944) Wettability of porous surfaces. Trans Faraday Soc 40:546–551
Wenzel RN (1936) Resistance of solid surfaces to wetting by water. Ind Eng Chem 28:988–994
Prince JA, Rana D, Singh G, Matsuura T, Jun Kai T, Shanmugasundaram TS (2014) Effect of hydrophobic surface modifying macromolecules on poly(vinylidene fluoride) membrane for direct contact membrane distillation. Chem Eng J 242:387–396
Savoji H, Rana D, Matsuura T, Tabe S, Feng C (2013) Development of plasma and/or chemically induced graft co-polymerized electrospun poly(vinylidene fluoride) membranes for solute separation. Sep Purif Technol 108:196–204
Prince JA, Singh G, Rana D, Matsuura T, Anbharasi V, Shanmugasundaram TS (2012) Preparation and characterization of highly hydrophobic poly(vinylidene fluoride)—clay nanocomposite electrospun nanofiber membranes for desalination using direct contact membrane distillation. J Membr Sci 397–398:80–86
Feng C, Khulbe KC, Matsuura T, Gopal R, Kaur S, Ramakrishna S, Khayet M (2008) Production of drinking water from saline water by air-gap membrane distillation using polyvinylidene fluoride nanofiber membrane. J Membr Sci 311:1–6
Feng C, Khulbe KC, Tabe S (2012) Volatile organic compound removal by membrane gas stripping using electro-spun nanofiber membrane. Desalination 287:98–102
Adamson AW, Gast AP (1997) Physical chemistry of surfaces, 6th edn. Wiley, New York
Webb PA, Orr C (1997) Analytical methods in fine particle technology. Micromeritics Instrument Corp, Norcross
Washburn EW (1921) The dynamics of capillary flow. Phys Rev 17:273–283
Dehl RE (1982) On the characterization of porosity in PTFE-carbon composite implant materials by mercury porosimetry. J Biomed Mater Res 16:715–719
Ryu YJ, Kim HY, Lee KH, Park HC, Lee DR (2003) Transport properties of electrospun nylon 6 nonwoven mats. Eur Polym J 39:1883–1889
Jena A, Gupta K (2002) Characterization of pore structure of filtration media. Fluid/Particle Sep J 14:227–241
Dickenson C (1992) Filters and filtration handbook, 3rd edn. Elsevier, Oxford
Hota G, Rajesh Kumar B, Ng WJ, Ramakrishna S (2008) Fabrication and characterization of a boehmite nanoparticle impregnated electrospun fiber membrane for removal of metal ions. J Mater Sci 43:212–217. doi:10.1007/s10853-007-2142-4
Xiao S, Hui M, Shen M, Wang S, Huang Q, Shi X (2011) Excellent copper (II) removal using zero-valent iron nanoparticle-immobilized hybrid electrospun polymer nanofibrous mats. Colloids Surf A 381:48–54
Sundarrajan S, Ramakrishna S (2007) Fabrication of nanocomposite membranes from nanofibers and nanoparticles for protection against chemical warfare stimulants. J Mater Sci 42:8400–8407. doi:10.1007/s10853-007-1786-4
Miao Y-E, Wang R, Chen D, Liu Z, Liu T (2012) Electrospun self-standing membrane of hierarchical SiO2@γ-AlOOH (Boehmite) core/sheath fibers for water remediation. ACS Appl Mater Interfaces 4:5353–5359
Royen PV, Schacht E, Ruys L, Vaeck LV (2006) Static secondary ion mass spectrometry for nanoscale analysis of electrospun nanofibres, surface characterisation. Rapid Commun Mass Spectrom 20:346–352
Acknowledgements
The authors gratefully acknowledge the financial support from the Environment and Water Industry (EWI) Development Council (Govt. of Singapore), the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Ministry of Environment, Ontario, Canada.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kaur, S., Sundarrajan, S., Rana, D. et al. Review: the characterization of electrospun nanofibrous liquid filtration membranes. J Mater Sci 49, 6143–6159 (2014). https://doi.org/10.1007/s10853-014-8308-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8308-y