
Abstract
A well organized calix[4]arene derivative in the cone configuration with carboxylic and phosphonic acid groups positioned at distal pairs has been prepared in order to investigate the extraction behavior of trivalent rare earths together with selected divalent metals. This product exhibited high extraction ability for the rare earths over the divalent metals investigated due to the complementarity of these trivalent ions with the above two functional groups without chelation to the phenoxy oxygen atoms. The extraction of the rare earths takes place by one of ion-exchange, where three protons are replaced by one Ln(III) cation. The stoichiometry of rare earth complexation was also determined by slope analysis, loading test and the continuous variation procedures. The extraction ability of the above reagent towards the respective rare earth ions was constant and their group separation over other metal ions proved possible. It was concluded that the presence of the two different functional groups on the calixarene structure results in a favorable synergistic interaction with the lanthanides. The results of stripping experiments for the loaded metals are also reported.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Calixarenes, cavity shaped macrocyclic phenolic oligomers, provide platforms of defined size and shape for binding guests. They have frequently been modified to obtain derivatives for use as ligands for binding metal ions. Several review articles have been published on calixarene ionophores [1–4]. These macrocyclic systems are attractive for use as host molecules because of their facile large-scale preparation, their ease of modification and the availability of different conformations and rim sizes. Their rigid cyclic structures and multifunctionality can lead to size discrimination behavior, a convergent effect reflecting their multiple (convergent) functional groups, as well as possible complementary effect of functional groups and/or allosteric effects [5]. The presence of a complementary group effect will enhance metal ion binding as shown in our previous works involving the extraction of rare earth [6] and sodium [7] ions with a tetraacetic acid derivative or the extraction of rare earth ions with phosphonic acid derivatives [8, 9]. It was also supported by our reports that calix[4]arene derivatives in their cone conformation incorporating two functional groups such as two long chain carboxylic acid or two acetic acid groups exhibited much less extraction ability for sodium than with four acetic acid groups [10, 11] and the system with two acetic acids and two alkyl branches also exhibited less extraction ability than that with four acetic acid groups [12].
In general, synergism is defined as two or more reagents working together to significantly enhance a given effect compared with the summation of their individual effects and was proposed by Blake et al. [13]. The concept was further discussed by Irving et al. [14]. Irving et al. pointed out that for a system involving a primary ion-exchangeable extraction reagent and a second neutral reagent, the first reagent acts to neutralize the charge on the metal ion while the second neutral reagent displaces the residual water molecules, consequently extraction efficiency is enhanced. Many examples of synergism in solvent extraction have been investigated and a number of reviews on extraction synergism have appeared [15–20]. Strzelbicki and Bartsch [21–23] prepared a range of lariat crown ethers [24] with ionizable groups on the arms. Subsequently, Umetani et al. proposed a further concept termed “intramolecular synergistic extraction” [25–28]. This involved multifunctional ligands, each incorporating an ion-exchangeable group and a crown ether ring. Since calixarenes are associated with multiple functional groups, they are suitable frameworks for use as intermolecular synergistic extraction reagents. In fact, many studies have been reported in which bifunctional calixarene compounds [29–34] (as well as calix crown compounds) were investigated [35–41], however, most of the former were not discussed in terms of intermolecular synergism. Nevertheless, calixarene-based derivatives are clearly promising compounds for investigating intermolecular synergistic effects.
As mentioned already, homo-functional calixarenes have been well demonstrated to show strong affinity for particular guest metal ions due to their binding complimentarily. Herein we report that the use of hetero-functional calixarenes containing different ionizable groups also lead to strong affinity with selected metal ions and, in particular, with ions from the rare earth group. Although the concept differs from that for conventional synergism, intermolecular synergism involving different (hetero-) binding groups also gives the prospect of yielding different ion selectivities relative to the situation where only homo binding groups are present. For example, tetrapropylenephosphonic acid derivatized calix[4]arene was shown to exhibit small heavier rare earth selectivity, while the tetracarboxylic acid derivative exhibited light to middle rare earth selectivity. Both reagents also exhibited high extraction efficiency of the trivalent rare earth metals over a selection of other metal ions. Thus, it was anticipated that calix[4]arene with two such functional groups would also exhibit separation efficiency for the rare earths over a range of other metal ions.
In the present work, we focus on the use of a new “cross” type calix[4]arene incorporating two propylenephosphonic and two acetic acid moieties for the group separation of rare earths via solvent extraction.
Experimental
Reagents
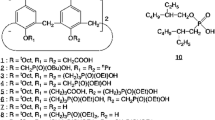
5,11,17,23-Tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene-25,26,27,28-tetrol (1) was synthesized in a similar manner to that described previously [6]. 25,27-Bis(ethoxycarbonylpropoxy)-26,28-dihydroxy-5,11,17,23-tetrakis-(1,1,3,3-tetramethylbutyl)calix[4]arene (2) was synthesized from 1 in a similar manner to that reported previously [10]. The reactant dibutyl 3-bromopropylphosphonate (3) was also prepared in a similar manner to that reported previously [8]. The present extraction reagent was prepared from 2 by diphosphonation with 3 at the remaining phenolic oxygen atoms and final hydrolysis with base as shown in Scheme 1. The product was identified as the desired one (in a cone conformation) by its 1H-NMR and IR spectra, TLC, and elemental analysis. For correlation with their extraction properties, the structure of 5 together with those of the reagents previously prepared, 25,26,27,28-tetrakis[3-{hydroxy(butyl)phosphoryloxy}propoxy]-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (6) and 25,26,27,28-tetrakis(carboxymethoxy)-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (7), are shown in Scheme 2.

Synthetic scheme of the present extraction reagent

Chemical structures of the present and previous extraction reagents
25,27-Bis(ethoxycarbonylpropoxy)-26,28-bis{3-(dibutylphosphoryloxy)propoxy}-tetrakis(1,1,3,3- tatramethylbutyl)calix[4]arene (4)
The diesterified compound 2 (0.305 g, 0.284 mmol), then sodium hydride (0.179 g, 62.7 % in oil, 4.68 mmol, 16 eq) were carefully added under a nitrogen stream to dry DMF (30 cm3) cooled in an ice bath. The mixture was stirred for an hour then the ice bath was removed and the solution was heated at 313 K for 4 h. Reactant 3 (0.477 g, 1.68 mmol, 6 eq) was added to the mixture which was then stirred for 24 h at room temperature. The reaction was monitored by TLC, and upon completion, the solvent was removed in vacuo. 1-Butanol (20 cm3) was added to deactivate excess sodium hydride (cooled in an ice bath). Solvent and excess 3 were removed using a rotary evaporator. The desired compound was extracted with chloroform from the residue and the organic solution was washed twice with both 1 M (M = mol dm−3) hydrochloric acid and distilled water. After drying over anhydrous magnesium sulfate, the solution was filtered and the solvent was then removed using a rotary evaporator to obtain a pale yellow viscous oily liquid. This pale yellow viscous liquid contained 3, together with some impurities. TLC yielded the main component (SiO2, chloroform:methanol = 20:1, R f = 0.53) and this crude compound was then hydrolyzed by multiple treatments with base in next step.
25,27-Bis(ethoxycarbonylpropoxy)-26,28-bis[3-{hydroxyl(butyl)phosphoryloxy}propoxy]-5,11,17,23-tetrakis(1,1,3,3-tetramethyl)butyl)calix[4]arene (5)
Under a nitrogen stream, crude 4 was dissolved in ethanol (10 cm3) and potassium hydroxide (0.269 g, 4.81 mmol) dissolved in water (0.900 g, 50.0 mmol) was mixed and refluxed for 50 h. The reaction was monitored by TLC, and upon completion, the solvent was removed on a rotary evaporator. The desired compound was extracted from the residue with chloroform. The chloroform solution was washed with 1 M hydrochloric acid (×3) and distilled water (×3). After drying over anhydrous magnesium sulfate, the mixture was filtered and the solvent was again removed on a rotary evaporator. The desired compound was recrystallized from acetonitrile. White powder 0.175 g (yield 57.4 % from 2); TLC (SiO2, chloroform:methanol = 4:1, R
f = 0.00–0.22); IR (KBr)  COO-H 2,500–3,300 cm−1 (br),
P(=O)O–H 2,690, 2,250, 1,650 cm−1 (br); 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) Δ 0.64 (18H, s, 2(CH3)3), 0.72 (18H, s, 2(CH3)3), 0.95 (18H, t + s, 2P-O(CH2)3CH
3 + 2(CH3)2), 1.20 (12H, s, 2(CH3)2), 1.40 (8H, m + s, 2P-O(CH2)2CH
2 + 2CH
2C(CH3)3), 1.65 (8H, s + m, 2CH
2C(CH3)3) + 2P-OCH2CH
2), 1.88 (4H, m, 2CH
2CH2COOH), 2.29 (8H, t + m, 2CH
2P + 2CH
2CH2P), 2.67 (4H, t, 2CH
2COOH), 3.12 (4H, d, 4exoCH2), 3.86 (8H, t + t, 4ArO-CH
2), 4.06 (4H, t, 2POCH
2), 4.30 (4H, d, 4endoCH2), 6.63 (4H, s, 2ArH(P)), 6.65 (4H, s(br), 2COOH + 2POOH), 6.85 (4H, s, 2ArH(C)). Found: C.70.70; H. 9.53 %, Calcd for C82H130O14P2:C, 70.26, H. 9.35 %.
COO-H 2,500–3,300 cm−1 (br),
P(=O)O–H 2,690, 2,250, 1,650 cm−1 (br); 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) Δ 0.64 (18H, s, 2(CH3)3), 0.72 (18H, s, 2(CH3)3), 0.95 (18H, t + s, 2P-O(CH2)3CH
3 + 2(CH3)2), 1.20 (12H, s, 2(CH3)2), 1.40 (8H, m + s, 2P-O(CH2)2CH
2 + 2CH
2C(CH3)3), 1.65 (8H, s + m, 2CH
2C(CH3)3) + 2P-OCH2CH
2), 1.88 (4H, m, 2CH
2CH2COOH), 2.29 (8H, t + m, 2CH
2P + 2CH
2CH2P), 2.67 (4H, t, 2CH
2COOH), 3.12 (4H, d, 4exoCH2), 3.86 (8H, t + t, 4ArO-CH
2), 4.06 (4H, t, 2POCH
2), 4.30 (4H, d, 4endoCH2), 6.63 (4H, s, 2ArH(P)), 6.65 (4H, s(br), 2COOH + 2POOH), 6.85 (4H, s, 2ArH(C)). Found: C.70.70; H. 9.53 %, Calcd for C82H130O14P2:C, 70.26, H. 9.35 %.
Other chemicals were purchased and used without further purification.
Distribution study
The extraction was performed in a similar manner to that described previously [8, 9]. An organic solution was prepared by diluting the extraction reagent 5 into analytical grade chloroform to yield a 5 mM concentration. Three aqueous solutions, each containing three rare earth ions, were prepared by dissolving analytical grade rare earth nitrates in 0.1 M hydrochloric acid or 0.1 M HEPES (2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid) to yield metal ion concentrations of 0.1 mM. The first solution contained La, Pr, and Nd, the second was Sm, Eu, and Gd, and the third Ho, Er and Y. The initial pH was adjusted by mixing the above two stock solutions. Equal volumes of the organic and aqueous phases were mixed and shaken vigorously at 303 K for more than 3 h. The metal concentrations and pH values of the aqueous solutions before and after shaking were measured by ICP-AES (Shimadzu ICPS-8100) and a pH meter (TOA-DKK HM-30R), respectively. The amount of the extracted metal ion was calculated from the difference between the metal concentration in the aqueous phase before and after shaking.
When using the continuous variation method (Job’s method) in the case of europium, the total concentration of the europium ion and the extraction reagent 5 was maintained at 1 mM. For the loading experiment, the europium concentration was increased to 5 mM. Both experiments were carried out in a similar manner to that described above. In the case of stripping, the organic phase (for which metal ions were quantitatively loaded in the forward extraction) was treated with fresh aqueous hydrochloric acid and the mixture shaken at 303 K for 12 h. After phase separation, in each case the metal concentration in the stripped aqueous solution was measured by ICP-AES.
For the extraction of divalent metal ions with 5, the aqueous solutions were prepared by dissolving all the metal nitrates in 0.1 M hydrochloric acid or 0.1 M HEPES buffer solution to such that the concentration of the mixed solutions was maintained at 0.1 mM. The procedures for the other metal ion systems were similar to those for rare earths.
Results and discussion
Distribution study
The extraction time to reach equilibration was investigated. The effect of shaking time on the extracted concentration of samarium ion with 5 is shown in Fig. 1. Although the test samples were mildly shaken at 130 rpm, the extraction rate was sufficiently rapid, with the extraction equilibration being reached within an hour.

Effect of shaking time on the extracted concentration of samarium ion with 5. [5] = 5 mM, [Sm3+] = 0.1 mM, initial pH = 1.60, 303 K
The pH dependence of the extraction of trivalent rare earths by 5 was investigated. The effect of the equilibrium pH on the distribution ratio D, of rare earth metals with 5 is shown in Fig. 2. The distribution ratio of the metal ion, D is the ratio of the extracted metal concentration in the organic phase to the metal concentration remaining in the aqueous phase at equilibrium, and is defined by Eq. (1),
where RE represents the trivalent rare earth metal.

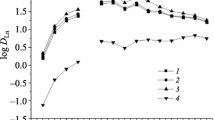
Effect of equilibrium pH on the distribution ratio of trivalent rare earth ions with 5. [5] = 5 mmol in chloroform, [metal ion] = 0.1 mmol dm−3 each in 0.1 M HCl – 0.1 M HEPES, shaking time = 6 h, 303 K
All plots shown in Fig. 2 are fitted by straight lines with a slope of 3, which corresponds to the charge on the respective rare earth metals. This result indicates that all trivalent metal ions had undergone ion exchange with three protons from the extraction reagent 5, even though the extraction took place in a low pH region. The selectivity order for 5 approximately favored the heavier to the lighter rare earths, which corresponds to the selectivity of conventional organophosphorus acid extractants, but not to carboxylic type extractants.
The pH dependence of selected metals with 5 was also investigated. The effect of equilibrium pH on the distribution ratio, D, for divalent metals with 5 is shown in Fig. 3. For comparison with the divalent ion data, data from Fig. 2 for trivalent erbium and lanthanum are also included; thus the extraction region between the plots for both these ions corresponds to that for all nine rare earths examined excluding the plots for the two trivalent ions, all plots shown in Fig. 3 are fitted by straight lines with a slope of 2, which also corresponds to the charge of the divalent metals. Reagent 5 appears useful not only for the separation of divalent metals, but also for group separation of rare earths over divalent metals, since the extraction pH region for the divalent metals is sufficiently higher than that for the rare earths. The selectivity order of 5 is rare earths > Pb > Zn > Cu > Ni. The metal selectivity for carboxylic acid functionalized extraction reagents is normally Pb(II) > Cu(II) > Zn(II) > rare earths > Ni(II) while for phosphonic acid derivatized extraction reagents the expected order is rare earths > Zn(II) > Pb(II) > Cu(II) > Ni(II). Thus, the selectivity of 5 for the rare earths and Zn(II) was attributed to the presence of phosphonic acid functionality and for Pb(II) and Cu(II) to the presence of carboxylic acid functionality.

Effect of equilibrium pH on the distribution ratio of divalent metal ions with 5. (Data of Er and La were taken from Fig. 2.) [5] = 5 mmol in chloroform, [metal ion] = 0.1 mmol dm−3 each in 0.1 M HCl – 0.1 M HEPES, shaking time = 6 h, 303 K
Loading experiments were carried out in order to confirm the stoichiometry of the interaction between the rare earth ions and 5, A typical result for the loading of Eu(III) by 5 is shown in Fig. 4. The ratio of the initial concentration of 5 to the loaded Eu(III) ion became close to two with increasing europium concentration. This result revealed that the stoichiometry of the rare earth ion complex of 5 was 1:2. Since the spacers attached to both functional group types are too long to promote chelate formation with phenoxy oxygen atoms for uptake of the rare earth ion, two pairs of different functional groups may not be sufficient to saturate the coordination number of the rare earth ion if the stoichiometry is 1:1. The phosphonic acid groups are preferable for complexing (by ion-exchange) rare earths rather than the carboxylic acid groups, however the coordination sphere will be unsaturated even by the presence of four phosphonic acid groups under 1:2 stoichiometry. This means that carboxylic acid groups will likely be employed for coordination at the unsaturated sites either by neutral carbonyl oxygen atoms or charged carboxylate groups.

Loading experiment of Eu(III) on 5. [5]initial = 5 mM, initial pH = 2.63 (0.1 M HCl – 0.1 M HEPES), shaking time = 6 h, 303 K
The continuous variation method was employed to obtain further confirmation of the interaction stoichiometry. A typical result is shown in Fig. 5 for the europium system. The extracted metal concentration in the organic phase reached a maximum at a mole fraction ratio between 0.6 and 0.7. The result supports a stoichiometry of 1:2. Three ion-exchangeable groups from among the four phosphonic and four carboxylic acid groups appear to act to extract this rare earth ion via the loss of three protons.

Job’s plots for Eu(III) with 5. [5]initial + [Eu3+]initial = 1 mM, initial pH = 2.63 (0.1 M HCl – 0.1 M HEPES), shaking time = 6 h, 303 K
The proposed extraction reaction is represented by Eq. (2),
where RE and H4L represent the rare earth metal and the extraction reagent 5, respectively, and K ex is the extraction equilibrium constant as represented by Eq. (3),
Since the distribution ratio was defined in Eq. (1), Eq. (3), this can be rewritten as Eq. (4),
After taking logarithms, Eq. (4) becomes Eq. (5),
Here the initial concentration of 5 is very much higher than the initial metal concentration while the difference in the extractant concentration before and after the extraction is negligible. Consequently, the latter two terms in Eq. (5) become essentially constant and the value of log D is proportional to the pH values. The result shown in Fig. 2 are in accord with Eq. (5), because the plots of log D versus pH lay on the straight lines with slopes of 3. Although stoichiometric data for the divalent metals were not obtained, since the plots of log D versus pH lie on straight lines with slopes of 2, clearly the extraction also took place by a simple two proton ion-exchange mechanism.
Complementarity of the phosphonic and carboxylic acid bifunctionality
In order to estimate the extraction ability and separation efficiency of 5 for the rare earth ions, half pH values, pH1/2, for the respective lanthanide ion systems were determined and the difference between the values for individual light and heavy metals obtained. The extraction equilibrium constants, K ex, and the separation factors, β for the extraction of individual rare earths with 5 were estimated. The values are listed in Table 1. The half pH value, pH1/2, is defined as the pH value that corresponds half metal extraction under the present experimental condition. The difference in half pH values, −ΔpH1/2 is calculated from the difference in the pH1/2 values for specific heavier and lighter metal ions (the negative ΔpH1/2 value indicates that the heavier metal ions are more easily extracted compared to the lighter metal ions). The separation factor, β, is defined by Eq. (6),
Here the extraction behavior for all the rare earths with 5 was the same and pH1/2 values obtained for the present and the previously reported extraction reagents were employed to compare the extraction ability and separation efficiency, together with the role of the two different functional groups on the rare earth extraction. For simple visual comparison, the relationship between pH1/2 and the reciprocal of the ionic radii (r−1) for trivalent six-coordinate rare earth metals is shown in Fig. 6 together with previous results for the tetrapropylenephosphonic acid derivative 6 [8] and the tetraacetic acid derivative 7 [6]. The y-axis is inverted so that the plots exhibiting high extraction ability appear upside down. The x-axis shows a lighter to heavier sequence of rare earths. As expected, the plots for 5 lie between those for the tetraphosphonic acid 6 and the tetracarboxylic acid 7 reagents, in accord with 5 containing both functional group types. That is, the extraction ability of 5 is weaker than that of 6 but is stronger than that of 7. Although 5 shows selectivity for heavier rare earths, the slope of the plot is almost flat, indicating that the separation efficiency of 5 is very poor. As discussed above, a single molecule of 5 is unable to saturate the coordination number of a rare earth ion, while the phosphonic acid functionality exhibits higher affinity than the carboxylic acid for heavier rare earth selectivity. Although the actual coordination number of rare earth ions was not clarified in this work, it is readily proposed that three of the functional groups are involved in ion-exchange while the others also coordinate via their carbonyl oxygen atoms. That is, three phosphonic acid groups from the four available were used for ion-exchange, with some or all of the remaining phosphonic/carboxylic acid groups also coordinating. This is in keeping with the extraction ability of 5 being closer to that of 6 rather than to that of 7. It is also noted that 5 and 6 with longer spacers and which cannot chelate, exhibited very high extraction compared to 7 (which incorporates shorter spacers that will allow chelation). The high extraction of 5 and 6 is attributed to the complementarity of the different functional groups with the same spacer length. The same concept can be extended to the complementarity exhibited by the different extractants shown in Fig. 7. That is, the extraction ability of 5 lies between the extraction abilities of 6 and 7, although it is closer to that of 6, because the phosphonic acid groups were primary to ion-exchange. (Strictly speaking, for a valid comparison the tetracarboxylic acid functionalized 7 should not be a tetraacetic acid derivative, but rather a tetrabutylic acid derivative, so that the space lengths would be the same.) The uptake of heavier lanthanide ions, which interact more weakly with the carboxylic acid groups, seem to be compensated by the interaction with the phosphonic acid groups. This proposal is also strongly supported by the selectivity pattern for the divalent metals shown in Fig. 3 as described above. That is, the selectivity of 5 for Zn(II) was attributed to the presence of phosphonic acid functionality and for Pb(II) and Cu(II) to the presence of carboxylic acid functionality. Reagent 5 is associated with hetero-functionality and yielded different metal selectivity to that obtained for the homo-functional 6 and 7, with total separation efficiency also suppressed due to the complementarily enhanced extraction efficiency by the different functional groups, e.g. the lead selectivity of 5 was remarkably suppressed compared with 7 [42]. For this reason, the intramolecular synergism with the different functionality enhances the complementarity by the different selectivity to balance the extraction ability, but offset respective selectivity of two functionalities for metals. Consequently although the extraction reagent 5 with bifunctionality of phosphonic and carboxylic acids exhibited poor selectivity to rare earth, it provided the possibility for group separation of rare earths over other metal ions.

Relationship between pH1/2 values and the reciprocal of ionic radii for trivalent rare earth metals, r−1. Closed triangle 5, open circle tetrapropylenephosphonic acid derivative, open square tetraacetic acid derivative of calix[4]arene

Different tendencies for the extraction of rare earths with 5 broken line, 6 plain line, 7 bold line
Stripping experiments
In a hydrometallurgical process, stripping is important for recovery of the desired metals and for regeneration of the extraction reagent. Since 5 exhibited similar extractability for all rare earths investigated, there is an advantage to simultaneously strip all of them in a single treatment. The effect of hydrochloric acid concentration on the percentage stripping of the rare earth metal ions loaded on 5 was investigated. La, Sm and Ho were selected as representatives of the light, middle and heavy rare earths. Since the extraction ability of 5 is high; 0.1 M hydrochloric acid was not sufficient to completely strip these loaded rare earths, while 0.5 M acid was effective for for stripping them. However, hydrochloric acid of the concentration higher 0.5 M led to lower stripping efficiency. A similar result was observed in our previous study on the stripping of the rare earths loaded on the tetracarboxylic acid derivative 7 [6]. It was also attributed to a different extraction mechanism; namely, not ion-exchange behavior by the phosphonic acid groups but rather coordination with phosphoryl oxygen atoms since the chloride concentration was very high and this ion acted as the counter anion (Fig. 8).

Effect of hydrochloric acid concentration on percentage stripping of rare earth metal ions loaded on the extraction reagent 5
Conclusions
A bifunctional cross-type of calix[4]arene derivative in a cone conformation has been prepared in order to investigate the extraction behavior of trivalent rare earths. Two different functionalities, phosphonic acid and carboxylic acid, on the calixarene acted in a complementary manner for the uptake of rare earth ions, although the ability to effect individual separation of rare earths was poor. However, the extraction ability of all the rare earths investigated with the new extraction reagent was effectively constant and their group separation over other (divalent) metal ions was demonstrated. Such complementarity based on metal ions interaction with different functional groups, each of which shows different selectivity provides intramolecular synergism to average the total affinity of a group of the guests with host.
References
Gutsche, C.D. (ed.): Calixarenes Revisited. Royal society of chemistry, Cambridge (1996)
Mandolini, L., Ungro, R. (eds.): Calixarenes in Action. Imperial college press, London (2000)
Lumetta, G.J., Rogers, R.D., Gopalan, A.S. (eds.): Calixarenes for Separations. ACS symposium series, vol. 757. ACS, Washington DC (2000)
Ludwig, R.: Calixarenes in analytical and separation chemistry. Fresenius J. Anal. Chem. 367(2), 103–128 (2000)
Ohto, K.: Molecular design and metal extraction behavior of calixarene compounds as host extractants. Ion Exch. Solvent Extr. 21, 8–127 (2013)
Ohto, K., Yano, M., Inoue, K., Yamamoto, T., Goto, M., Nakashio, F., Shinkai, S., Nagasaki, T.: Solvent extraction of trivalent rare earth metal ions with carboxylate derivatives of calixarenes. Anal. Sci. 11(6), 893–902 (1995)
Ohto, K., Ishibashi, H., Kawakita, H., Inoue, K., Oshima, T.: Allosteric coextraction of sodium and metal ions with calix[4]arene derivatives 1. Role of the first-extracted sodium ion as an allosteric trigger for self-coextraction of sodium ions with calix[4]arene tetracarboxylic acid. J. Incl. Phenom. Macrocycl. Chem. 65, 111–120 (2009)
Ohto, K., Matsufuji, T., Yoneyama, T., Tanaka, M., Kawakita, H., Oshima, T.: Preorganized, cone-conformational calix[4]arene possessing four propylenephosphonic acids with high extraction ability and separation efficiency for trivalent rare earth elements. J. Incl. Phenom. Macrocycl. Chem. 71(3,4), 489–497 (2011)
Ohto, K., Takedomi, A., Chetry, A.B., Morisada, S., Kawakita, H., Oshima, T.: The effect of phenoxy oxygen atoms on extremely high extraction ability and less separation efficiency of trivalent rare earth elements with tetraphosphonic acid derivative of calix[4]arene. J. Incl. Phenom. Macrocycl. Chem. 77(1–4), 363–373 (2013)
Ohto, K., Shiratsuchi, K., Inoue, K., Goto, M., Nakashio, F., Shinkai, S., Nagasaki, T.: Extraction behavior of copper(II) ion by calixarene carboxylates derivatives preorganized by sodium ion. Solvent Extr. Ion Exch. 14(3), 459–478 (1996)
Yoneyama, T., Sadamatsu, H., Kuwata, S., Kawakita, H., Ohto, K.: Allosteric coextraction of sodium and metal ions with calix[4]arene derivatives 2: first numerical evaluation for the allosteric effect on alkali metal extraction with crossed carboxylicacidtypecalix[4]arenes. Talanta 88, 121–128 (2012)
Sadamatsu, H., Morisada, S., Kawakita, H., Ohto, K.: unpublished data
Blake, C.A., Baes Jr, C.F., Brown, K.B., Coleman, C.F., White, J.C.: Solvent extraction of uranium and other metals by acidic and neutral organophosphorus compounds. Proceedings of the United Nations International Conference on the Peaceful Uses of Atomic Energy 28, 289–298 (1958)
Irving, H., Edgington, D.N.: Synergic effects in the solvent extraction of the actinides. I. Uranium(VI). J. Inorg. Nucl. Chem. 15, 158–170 (1960)
Akaiwa, H., Kawamoto, H.: The application of synergistic extraction to analytical chemistry. Rev. Anal. Chem. 6(1), 65–86 (1982)
Dukov, I.L.: Synergistic solvent extraction of metals. Part 1. Chelating extractants plus amines or quaternary ammonium salts. Anal. Lab. 5(3), 147–161 (1996)
Dukov, I. L.: Synergistic solvent extraction of metals. Part 2. Alkylphosphoric and carboxylic acids and neutral organophosphorus extractants plus amines or quaternary ammonium salts. Anal. Lab. 5(4), 219-161 (1996)
Bond, A.H., Dietz, M.L., Chiarizia, R.: Incorporating size selectivity into synergistic solvent extraction: a review of crown ether-containing systems. Ind. Eng. Chem. Res. 39(10), 3442–3464 (2000)
Nishihama, S., Hirai, T., Komasawa, I.: Review of advanced liquid-liquid extraction systems for the separation of metal ions by a combination of conversion of the metal species with chemical reaction. Ind. Eng. Chem. Res. 40(14), 3085–3091 (2001)
Cox, M.: Liquid-liquid extraction and liquid membranes in the perspective of the twenty-first century. In: Aguilar, M., Cortina, J.L. (eds.) Solvent Extraction and Liquid Membranes, pp. 1–19. CRC Press, Boca Raton (2008)
Strzelbicki, J., Bartsch, R.A.: Extraction of alkali metal cations from aqueous solutions by a crown ether carboxylic acid. Anal. Chem. 53(12), 1894–1899 (1981)
Strzelbicki, J., Bartsch, R.A.: Solvent extraction of alkali metal cations from aqueous solutions by highly lipophilic crown ether carboxylic acids. Anal. Chem. 53(14), 2247–2250 (1981)
Strzelbicki, J., Bartsch, R.A.: Extraction of alkaline earth cations from aqueous solutions by crown ether carboxylic acids. Anal. Chem. 53(14), 2251–2253 (1981)
Gokel, G.W., Dishong, D.M., Diamond, C.J.: Lariat ethers. Synthesis and cation binding of macrocyclic polyethers possessing axially disposed secondary donor groups. J. Chem. Soc., Chem. Commun. 22, 1053–1054 (1980)
Umetani, S., Yamazaki, S., Ogura, K.: Solvent extraction of metal ions with novel 4-acyl-5-pyrazolones having crown ether moiety as intramolecular synergist. Proceedings of International Solvent Extraction Conference ISEC 2002, Cape Town, South Africa, 420–424 (2002)
Umetani, S., Ito, M., Shimojo, S., Kurahashi, K., Yamazaki, S., Ogura, K.: Solvent extraction of divalent transition metal ions with diaza-crown ethers having two acylpyrazolone moieties. Solvent Extr. Res. Dev., Jpn. 14, 177–181 (2007)
Kurahashi, K., Umetani, S., Sohrin, Y.: Solvent extraction of divalent metal ions with azacrown ether substituted acylpyrazolones. Anal. Sci. 24(2), 225–229 (2008)
Shimojo, K., Okamura, H., Hirayama, N., Umetani, S., Imura, H., Naganawa, H.: Cooperative intramolecular interaction of diazacrown ether bearing β-diketone fragments on an ionic liquid extraction system. Dalton Trans. 25, 4850–4852 (2009)
Ogata, M., Fujimoto, K., Shinkai, S.: Molecular design of calix[4]arene-based extractants which show high Ca2 + selectivity. J. Am. Chem. Soc. 116(10), 4505–4506 (1994)
Casnati, A., Fischer, C., Guardigli, M., Isernia, A., Manet, I., Sabbatini, N., Ungaro, R.: Synthesis of calix[4]arene receptors incorporating (2,2′-bipyridin-6-yl)methyl and (9-methyl-1,10-phenanthrolin-2-yl)methyl chromophores and luminescence of their Eu3 + and Tb3 + complexes. J. Chem. Soc., Perkin Trans. 2(3), 395–399 (1996)
Yaftian, M.R., Burgard, M., Matt, D., Wieser, C.: Dieleman, C: multifunctional calix[4]arenes containing pendant amide and phosphoryl groups: their use as extracting agents and carriers for alkali cations. J. Incl. Phenom. Mol. Recog. Chem. 27(2), 127–140 (1997)
Arnaud-Neu, F., Browne, J.K., Byrne, D., Marrs, D.J., McKervey, M.A., O’Hagan, P., Schwing-Weill, M.J., Walker, A.: Extraction and complexation of alkali, alkaline earth, and F-element cations by calixaryl phosphine oxides. Chem. Eur. J. 5(1), 175–186 (1999)
Beer, P.D., Drew, M.G.B., Hesek, D., Kan, M., Nicholson, G., Schmitt, P., Sheen, P.D., Williams, G.: A neutral uranyl dimeric complex and remarkable extraction properties of a 1-acid 3-diethyl amide substituted calix[4]arene ligand. J. Chem. Soc., Dalton Trans. 17, 2783–2786 (1998)
Mikulasek, L., Gruner, B., Dordea, C., Rudzevich, V., Boehmer, V., Haddaoui, J., Hubscher-Bruder, V., Arnaud-Nue, F., Caslavsky, J., Selucky, P.: Tert-butyl-calix[4]arenes substituted at the narrow rim with cobalt bis(dicarbollide)(1-) and CMPO groups - new and efficient extractants for lanthanides and actinides. Eur. J. Org. Chem. 28, 4772–4783 (2007)
Casnati, A., Ungaro, R., Asfari, Z., Vicens, J.: Crown ethers derived from calix[4]arenes. In: Asfari, Z., Boehmer, V., Harrowfield, J.M., Vicens, J. (eds.) Calixarenes 2001, pp. 365–384. Kluwer, Netherlands (2001)
Alfieri, C., Dradi, E., Pochini, A., Ungaro, R., Andreetti, G.D.: Synthesis and x-ray crystal and molecular structure of a novel macrobicyclic ligand: crowned p-tert-butylcalix[4]arene. J. Chem. Soc., Chem. Commun. 19, 1075–1077 (1983)
Ungaro, R., Pochini, A., Andreetti, G.D.: New ionizable ligands from p-tert-butylcalix[4]arene. J. Incl. Phenom. Macrocycl. Chem. 2(1–2), 199–206 (1984)
Casnati, A., Pochini, A., Ungaro, R., Ugozzoli, F., Arnaud, F., Fanni, S., Schwing, M.-J., Egberink, R.J.M., de Jong, F., Reinhoudt, D.N.: Synthesis, complexation, and membrane transport studies of 1,3-alternate calix[4]arene-crown-6 conformers: a new class of cesium selective ionophores. J. Am. Chem. Soc. 117(10), 2767–2777 (1995)
Thuery, P., Nierlich, M., Lamare, V., Dozol, J.-F., Asfari, Z., Vicens, J.: Bis(crown ether) and azobenzocrown derivatives of calix[4]arene. A review of structural information from crystallographic and modelling studies. J. Incl. Phenom. Macrocycl. Chem. 36(4), 375–408 (2000)
Haverlock, T.J., Bonnesen, P.V., Sachleben, R.A., Moyer, B.A.: Analysis of equilibria in the extraction of cesium nitrate by calix[4]arene-bis(t-octylbenzo-crown-6) in 1,2-dichloroethane. J. Incl. Phenom. Macrocycl. Chem. 36(1), 21–37 (2000)
Ramachandran, B.R., Baker, S.D., Suravajhula, G., Derosa, P.A.: Selective complexation of alkali metal ions using crown ethers derived from calix[4]arenes: a computational investigation of the structural and energetic factors. J. Incl. Phenom. Macrocycl. Chem. 75(1–2), 185–195 (2013)
Ohto, K., Fujimoto, Y., Inoue, K.: Stepwise extraction of two lead ions with a single molecule of calix[4]arene tetracarboxylic acid. Anal. Chim. Acta 387(1), 61–69 (1999)
Acknowledgments
This work was financially supported in part by a Grant-in-Aid for Scientific Research No. 25410192 from the Ministry of Education, Science, and Culture.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chetry, A.B., Matsufuji, T., Adhikari, B.B. et al. Intramolecular synergism for group separation extraction of trivalent rare earths by a cross type calix[4]arene with phosphonic and carboxylic acid bifunctionality. J Incl Phenom Macrocycl Chem 81, 301–310 (2015). https://doi.org/10.1007/s10847-014-0457-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-014-0457-8