
Abstract
A cone confomational p-t-octylcalix[4]arene with four methylenephosphonic acids at the lower rim as well as its monomeric analog have been synthesized as a new extraction reagent to investigate the extraction behavior of the nine trivalent rare earth elements: Y, La, Pr, Nd, Sm, Eu, Gd, Ho and Er. The new calix[4]arene exhibited significantly higher extraction capacity than not only the monomeric derivative and the commercial extraction reagent, 2-ethylhexyl hydrogen 2-ethylhexylphosphonate, but also the cone conformational calix[4]arene extractants employed in our previous work, composed of a tetrapropylenephosphonic acid with a longer spacer, a tetraphosphonic acid at the upper rim, and tetraacetic acid at the lower rim. A dependence on the pH was observed for the new extractant in the highly acidic region, and the extraction took place via a simple ion-exchange mechanism. Using slope analysis, the stoichiometries of tetrameric and monomeric extractants to rare earth metal ions were determined to be 2:1 and 3:1, respectively. Using the proposed extraction equations, extraction equilibrium constants and separation factors were estimated. The extremely high extraction ability and moderate separation efficiency were attributed to the chelating effect of the phosphonic acid and the phenoxy oxygen atom. The effect of the phenoxy oxygen atom on extraction ability and separation efficiency of calix[4]arene derivatives was found to be significant.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rare earth metals are difficult to mutually separate due to their similar chemical properties and ionic size. These metals are key elements in a number of advanced industrial applications, including magnetic materials, luminescent materials, superconductors, catalysts, polishers, and lasers. Separation techniques are necessary because high purity is generally required for these applications. Among the many techniques for recovery and separation of metals, solvent extraction has been used extensively due to its high separation efficiency, excellent versatility, and mass disposal potential. The role of the extraction reagents is very important in the solvent extraction method. Various types of extractants have been prepared, and organophosphorus acid materials were found to be effective for rare earth extractions [1]. Peppard et al. [2–6] performed extensive investigations on the solvent extraction of rare earth elements using organophosphorus extractants for the application of separation from nuclear waste and actinides. Yuan et al. [7–10] prepared a series of acidic phosphorus esters to study the relationship between structure and extraction behavior. Furthermore, novel organophosphorus acid extraction reagents for rare earth elements have also been prepared [11–14].
Calixarenes are macrocyclic oligomers that provide three-dimensional coordination sites for a variety of interesting compounds [15, 16]. The ability to recognize and discriminate metal ions is a remarkable feature of these site-specific receptors. Several review articles have been published on these interesting ionophores [17–25]. Acidic organophosphorus compounds based on a calixarene framework for rare earth elements have been reported. Oshima et al. [26] prepared hexaphosphorylated calix[6]arene to investigate the extraction behavior of nine rare earth elements and reported high extraction ability and selectivity for heavy rare earth metals. Jurecka et al. [27] prepared three calix[4]arene derivatives containing two dibasic phosphonic acids with different spacer lengths in a cone conformation and also calix[4]arene derivative with four dibasic phosphonic acids and a propylene spacer length in a cone conformation [28]. They found that the extraction efficiencies decrease with increasing length of the CH2 spacer. The tetraphosphonic acid with a propylene spacer length exhibited selectivity for light rare earth metals, whereas the three diphosphonic acid derivatives demonstrated selectivity for heavy rare earth metals. In addition, calix[4]arene derivatives with four phosphonic acids at upper rim were prepared in our previous work [29, 30]. Several phosphorus-containing calixarenes have been prepared [31–35]. Since the binding site at the upper rim is much larger than the ion diameter of rare earth metals, the separation efficiency for phosphorus-containing calixarenes at upper rim may be poorer than that of the corresponding monomeric analogue, because acidic phosphorus groups tend to smaller heavier rare earth selectivity. Thus, the effect of the cyclic structure should well multiply by the effect based on the functional group affinity [29].
More recently, calix[4]arene derivatives containing four mono-basic propylenephosphonic acid groups at the lower rim were prepared to investigate the extraction behavior of rare earth metals including both the extraction ability and the mutual separation efficiency [36]. These reagents exhibited extremely high extraction efficiency for trivalent rare earth metals and significant heavy rare earth selectivity.
In this contribution, we have synthesized calix[4]arene derivatives containing four mono-basic methylenephosphonate groups at the lower rim to investigate the extraction behavior of rare earth metals (Y, La, Pr, Nd, Sm, Eu, Gd, Ho, and Er), including both the extraction ability and the mutual separation efficiency. By comparing the extraction efficiency of two monomers, that is, the corresponding monomer and a phosphonic acid derivative with its similar lipophilicity and without phenoxy oxygen atoms, it is indirectly interpreted that the extremely high extraction ability of the calix[4]arene extractant is attributed to the chelation of the phosphonic acid and the phenoxy oxygen atom. Although the chelation contributes to the high extraction ability, it also results in poor separation efficiency.
Experimental
Reagents
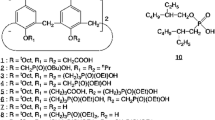
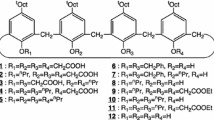
The chemical structures of extractant 5 and the corresponding monomeric analog 7 together with the calix[4]arene phosphonic acid derivative with a longer spacer 8, its monomeric acid derivative 9, carboxylic acid derivatives of calix[4]arene 10, and the commercial extractant 11 (PC-88A is its commercial name), are shown in Fig. 1.

Chemical structures of extractants
The commercial extractant 11, 2-ethylhexyl 2-ethylhexylphosphonic acid was kindly supplied by the Daihachi Chemical Industry Co., Ltd. and was used without further purification. The other chemicals were purchased and also used without further purification. The extractant 5 has been prepared in three steps, which include cyclization, tetraphosphonation, and basic hydrolysis from the corresponding phenol. 5,11,17,23-Tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene-25,26,27,28-tetol 3 was synthesized in a similar manner to a previously described procedure [37]. The synthetic schemes for extractants 5 and 7 are shown in Fig. 2.

Synthetic schemes for extractants 5 and 7
Dibutyl hydroxymethylphosphonate(1) [38]
Under a nitrogen stream, dibutylphosphite (49.7 g, 256 mmol) was added to paraformaldehyde (9.28 g, 309 mmol) and dry potassium carbonate (1.80 g, 13.0 mmol) in 100 cm3 1-butanol. The mixture was stirred and heated at 135 °C for 30 h. The solvent was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed with 1 M (M = mol dm−3) hydrochloric acid, saturated brine and distilled water each three times. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo and a colorless liquid was obtained. 42.3 g (yield 73.7 %), 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) δ 0.93 (6H, t, 2CH3), 1.40 (4H, m, 2CH 2 CH3), 1.67 (4H, m, 2CH 2 CH2CH3), 3.92 (2H, d, J = 6.0 Hz, CH2OH), 4.08 (4H, t, OCH2), 4.67 (1H, br s, OH).
Dibutyl phosphonomethyl tosylate (2) [39]
Under a nitrogen stream, 1 (30.0 g, 133 mmol) was added to 150 cm3 dry pyridine in an ice bath. To this mixture, p-tosyl chloride (32.6 g, 171 mmol) was added and stirred for 24 h. The reaction was monitored by TLC, and upon completion, pyridine was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed with 1 M hydrochloric acid, saturated brine and distilled water each three times. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo and the residue was purified by column chromatography (SiO2, chloroform:hexane = 5:1 v/v and chloroform); yellow viscous liquid; 26.3 g (yield 51.8 %); 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) δ 0.92 (6H, t, 2 CH2 CH 3 ), 1.37 (4H, m, 2CH 2 CH3), 1.65 (4H, m, 2CH 2 CH2CH3), 2.46 (3H, t, CH3), 4.06 (4H, t, POCH2), 4.19 (2H, d, J = 10.2 Hz, PCH2O), 7.36 (2H, d, ArH-CH3), 7.80 (2H, d, ArH-S).
25,26,27,28-Tetrakis(butyl hydrogen phosphoryloxy)methoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (5) [27]
Under a nitrogen stream, the parent calix[4]arene 3 (10.0 g, 11.5 mmol), potassium carbonate (31.3 g, 226 mmol), and 2 (31.4 g, 82.9 mmol) were added to 100 cm3 dry acetonitrile and the mixture was refluxed for 144 h. The reaction was monitored by TLC, and upon completion, the solvent was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed with 1 M hydrochloric acid and distilled water each three times. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo and the residue was purified by column chromatography (SiO2, chloroform and chloroform:butanol = 5:1 v/v); yellow viscous liquid; 7.02 g. Although the crude compound was mainly composed of the desired compound 4, 25,26,27,28-tetrakis(dibutylphosphoryloxy)methoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene, it also contained the reactant 2 as an impurity. Nevertheless, hydrolysis was performed on the crude material as described below.
Under a nitrogen stream, the crude 4 (2.03 g) was dissolved in 50 cm3 ethanol. To the mixture, potassium hydroxide (2.08 g, 37.1 mmol) in water (28.9 g, 1.61 mol) was added. The mixture was refluxed for 120 h. The reaction was monitored by TLC, and upon completion, the solvent was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed once with 6 M and then with 1 M hydrochloric acid, followed by four washes with distilled water. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo. The desired compound was recrystallized from acetonitrile and chloroform; white powder; 0.792 g (yield 44.8 % from 3); IR (KBr) disappeared νO–H 3,150 cm−1 (br), νP(O)OH 2,671, 2,285 and 1,671 cm−1; 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) 0.69 (36H, s, C(CH3)3), 0.90 (12H, t, CH2 CH 3 ), 1.10 (24H, s, C(CH3)2), 1.36 (8H, m, CH 2 CH3), 1.58 (8H + 8H, s + m, CCH2C + OCH2 CH 2 ), 3.24 (4H, d, exo-CH2), 3.92 (8H, m, OCH 2 CH2), 4.50 (8H, d, J = 12.3 Hz, CH2P), 4.63 (4H, d, endo-CH2), 6.80 (8H, s, ArH), 10.50 (4H, s(br), OH), Found: C, 64.93; H, 8.97 %, Calcd for C80H132O16P4: C, 65.20, H, 9.03 %.
Butyl hydrogen[4-(1,1,3,3-tetramethylbutyl)phenoxymethyl]phosphonate (7)
Under a nitrogen stream, p-t-octylphenol (9.99 g, 48.0 mmol), potassium carbonate (35.1 g, 254 mmol), and 2 (26.0 g, 69.0 mmol) were added to 100 cm3 dry acetonitrile and the mixture was refluxed for 90 h. The reaction was monitored by TLC, and upon completion, the solvent was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed with 1 M hydrochloric acid and distilled water each three times. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo; yellow viscous liquid; 28.1 g. Although the crude compound was mainly composed of the desired compound 6, dibutyl[4-(1,1,3,3-tetramethylbutyl)phenoxymethyl]phosphonate, it also contained the reactant 2 as an impurity. Nevertheless, hydrolysis was performed on the crude material as described below.
Under a nitrogen stream, the crude 6 (12.0 g) was dissolved in 100 cm3 ethanol. To this mixture, potassium hydroxide (14.1 g, 249 mmol) in water (50.3 g, 2.79 mol) was added. The mixture was refluxed for 36 h. The reaction was monitored by TLC, and upon completion, the solvent was removed in vacuo. Chloroform was added to the residue to extract the desired compound. The organic solution was washed with 1 M hydrochloric acid and distilled water each three times. After drying over anhydrous magnesium sulfate, the solvent was removed in vacuo; yellow viscous liquid; 4.12 g (yield 54.6 % from p-t-octylphenol); IR (neat) νP(O)OH 2,627, 2,278 and 1,678 cm−1; 1H-NMR (300 MHz, CDCl3, TMS, 30 °C) 0.70 (9H, s, C(CH3)3), 0.91 (3H, t, CH2 CH 3 ), 1.41 (6H + 2H, s + m, C(CH3)2 + CH 2 CH3), 1.64 (2H + 2H, t + s, OCH2 CH 2 + CCH2C), 4.15 (8H, m, OCH 2 CH2), 4.27 (8H, d, J = 10.8 Hz, CH2P), 6.85 (2H, d, O-ArH), 7.26 (2H, d, C-ArH), 11.82(1H, s(br), OH), Found: C, 63.43; H, 9.27 %, Calcd for C19H33O4P: C, 64.02, H, 9.33 %.
Distribution study of rare earth metals
The extraction was performed in a similar manner as previously described [36, 37]. An organic solution was prepared by diluting each extractant with analytical grade of chloroform to the concentration of 5.0 mM for 5 and 20 mM for 7. The three types of aqueous solutions, each containing three types of rare earth ions, were prepared by dissolving analytical grade rare earth chloride in 0.01, 0.1, 1.0 or 5.0 M hydrochloric acid and then diluting to a concentration of 0.1 mM. Among 17 rare earth elements, some pairs of adjacent elements are remarkably difficult to be mutually separated such as Pr/Nd, Sm/Eu, Eu/Gd, Ho/Er, Ho/(Y)/Er in using organophosphorus acid reagents. Nine elements containing above pairs were selected for this experiment in similar manner as the previous one. The first solution contained La, Pr, and Nd; the second Sm, Eu, and Gd; and the last Ho, Er, and Y. In the case of the monomeric extractant, aqueous solutions were prepared by dissolving analytical grade rare earth chloride in 0.1 M hydrochloric acid and 0.1 M HEPES (2-[4-(2-hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) buffer. The hydrochloric acid concentration or the initial pH value of the aqueous solution was adjusted by arbitrarily mixing above stock solutions. Equal volumes (5 cm3) of both phases were mixed and vigorously shaken at 303 K for more than 3 h. After phase separation, the metal concentrations in the aqueous solutions were measured by ICP-AES (Shimadzu ICPS-8100). The extracted metal ion concentrations were calculated from the differences in metal concentrations of the aqueous phase before and after equilibration. Proton concentrations were calculated from the mixing ratio of hydrochloric acid solutions, and the proton activities were estimated using activity coefficients [40]. The equilibrium pH was assumed to be the same as the initial pH because the pH was low enough to ignore the change before and after the extraction. When using the continuous variation method (Job’s method), the total concentration of europium ion and extractant was maintained at 1 mM.
Results and discussion
First, the extraction time was first investigated. The effects of shaking time on the extraction percentage of trivalent samarium, europium, and gadolinium ions with 5 and 7 are shown in Fig. 3a, b. Although the extraction samples were shaken mildly at 130 rpm, the extraction of rare earth metal ions was fast and reached equilibrium within an hour for 5 and 15 min for 7. The equilibrium time was longer for the calix[4]arene compound than the corresponding monomeric acid, perhaps because of the strong intramolecular hydrogen bonding of the former compound due to its localized polarity and cone conformation.

Effects of shaking time on the extraction percentage of trivalent samarium, europium, and gadolinium ions. a [5] = 5 mM, initial pH = 0.31; b [7] = 20 mM, initial pH = 1.53
Next, the pH dependence of trivalent rare earths with the extractants was investigated. The effects of pH on the distribution ratio of rare earth metals with 5 and 7 are shown in Fig. 4a, b. The pH region for both extraction reagents was remarkably low, meaning that both extraction reagents exhibited high extraction ability for rare earths, and the extraction occurred despite a high proton concentration. All points in Fig. 4 can be fit by straight lines with a slope of 3, which is equivalent to the charge of rare earth metals. This result indicates that all trivalent metal ions are ion-exchanged with the three protons from the extractants, even in such a low pH environment. The following extraction selectivity was observed: Er > Y > Ho > Gd > Eu > Sm > Nd > Pr > La. Heavier rare earth metals are preferentially extracted over lighter ones. The selectivity trends are similar to those of conventional acidic phosphorus extractants, although the selectivity is relatively poor. For acidic phosphorus extractants, yttrium is placed between holmium and erbium [41].

Effects of pH on the distribution ratio of rare earth metals with a 5 and b 7
Another experiment was performed using Job’s method to confirm the stoichiometry of the extraction reaction. A representative Job’s plots for Eu(III) with 5 and 7 are shown in Fig. 5a, b, respectively. The maximum ratio of extractant concentration to total concentration of extractant and europium ion is 0.53 for 5 and 0.75 for 7. These results indicated that the trivalent europium ion formed a 1:1 (metal:extraction reagent) complex with 5, and a 1:3 complex with 7.

Job’s plots for Eu(III) with a 5 and b 7. [Extractant]initial + [Eu ion] initial = 1 mM, initial pH = 5.30
It should be noted that the stoichiometries determined by the slope analysis of the extractant concentration dependence are different from those derived by Job’s method. The effects of extractant concentration on the distribution ratio of samarium and europium ions with 5 and 7 are shown in Fig. 6a, b. All points for 5 in Fig. 6a can be fit by straight lines with the slope of 2, whereas those of 7 in Fig. 6b exhibit slopes of 3. These results indicate that the stoichiometries of samarium and europium ions with 5 and 7 are 1:2 and 1:3 (metal:extraction reagent), respectively, which is inconsistent with the result obtained using Job’s method. The difference in the stoichiometry values for 5 determined by the two methods is caused by the different experimental conditions. That is, the extractant concentration in Job’s method is equivalent to the metal concentration at maximum point, whereas in the slope analysis method, it is much higher than that of the metal. Accordingly, extractant 5 forms 1:2 complexes with rare earth metals when the extractant concentration exceeds that of the metal. Thus, the stoichiometry for the pH dependence experiments was presumed to be 1:2. In this case, some of oxygen atoms of the functional groups may be related to the complexation so as to saturate the coordination number of rare earth elements.

Effects of the extractant concentration on the distribution ratio of Sm and Eu ions with a 5 (initial pH = 0.08) and b 7 (initial pH = 1.17)
Based on the above results and presumptions, the stoichiometric relationship in the extraction reaction of trivalent rare earths with 5 is proposed by the Eq. (1);
where RE and H4L represent the rare earth metal and tetrameric phosphonic acid 5, respectively, and K ex is the extraction equilibrium constant. This stoichiometric relationship yields the following equilibrium relationship:
The Eq. (2) can be rewritten as Eq. (3):
where D value is the distribution ratio defined by Eq. (4).
where subscripts org and aq represent the species in the organic and aqueous phases, respectively.
The slope of the pH dependence in Fig. 4a is 3, which is derived from the charge of the rare earth elements. Because the initial extractant concentration is significantly higher than the total metal concentration, the difference in the extractant concentration before and after the extraction is negligible. Consequently, the results of the pH dependence study are in agreement with the extraction reaction (1).
The slope of the extractant concentration dependence in Fig. 6a is 2, which is consistent with the stoichiometry of the complex. The results of the extractant concentration dependence are also in agreement with the Eq. (1).
Similarly, the extraction relation of trivalent rare earths with 7 is proposed by the Eq. (5);
where HL represents the monomeric phosphonic acid 7. The Eq. (5) can be rewritten as Eq. (6):
The results shown in Figs. 4b and 6b are also consistent with the extraction Eq. (5).
Using the above data and equations, the half pH values (pH1/2), the difference between the half pH values (−ΔpH1/2), the extraction equilibrium constants (K ex) and the separation factors (β), were estimated for each extractant. The half pH value, pH1/2, is defined as the pH value at which half of the metal ions are extracted under the experimental conditions used in this study. The difference between the half pH values, −ΔpH1/2, is calculated from the difference in the pH1/2 values between the heavier and lighter metal ions (a negative ΔpH1/2 value means that a heavier metal ion is more easily extracted in comparison with a lighter one). The separation factor, β, is defined by Eq. (7):
The obtained values are provided in Table 1, together with the values for tetraphosphonic acid calix[4]arene with a longer spacer 8 [36], and the commercial extractant 11 [36]. The K ex values for the extractants are not directly comparable due to the difference in stoichiometries, whereas the pH1/2, −ΔpH1/2, and β values are directly comparable.
Figure 7 provides a visual comparison of the relationship between the pH1/2 values and the reciprocal number of ionic radii for trivalent rare earth metals with a coordination number of six, r−1. The y-axis is inverted such that the plots for the extractants with high extraction efficiency appear upside down. Although the stoichiometry for each extractant is not the same, the extraction ability and separation efficiency can be compared because the number of functional groups for all experimental conditions is the same. The extraction ability of extractant 5 is much greater than that of 7, 8, 10 and 11, because its plots are clearly higher than those of other extractants, which is attributed to the highly pre-organized structure for the rare earth metals due to the size-fitting effect, the complimentary effect and the converging effect of the four phosphonic acids arranged in a cone conformation. The difference between the plots for each rare earth ion corresponds to the separation efficiency. Although the plots for the extractants do not exactly form straight lines and appear to organize as ‘tetrads’ [42], the slope between the two plots roughly indicates the metal selectivity and separation efficiency. As described earlier, 5 exhibits high heavy rare earth metal selectivity with an extraction selectivity of Er > Y > Ho > Gd > Eu > Sm > Nd > Pr > La.

Relationship between pH1/2 values and the reciprocal number of ionic radii for trivalent rare earth metals, r−1
The separation efficiency of 5 is comparable to that of the monomeric analog 7 lower than that of longer spacer derivative 8 and the commercial extractant 11, and higher than that of the tetraacetic acid 10. Calix[4]arene derivatives and its corresponding monomers exhibited higher extraction ability and separation efficiency compared with the calix[4]arene tetraacetic acid materials. Molecular design, including the nature of the functional group, is shown to be truly significant.
Extractant 5 exhibited extremely high extraction ability among the extractants introduced in this work, but only moderate separation efficiency. Why did it exhibit much higher extraction ability and lower separation efficiency than derivative 8 with a longer spacer and the commercial extractant 11? To explain this phenomenon, the results for the monomeric derivative 7 must be considered. The extraction efficiency of 7 is much higher than that of tetrapropylenephosphonic acid calix[4]arene 8. Although the monomeric derivative 7 does not correspond to the calix[4]arene derivative 8, the result is unusual because calix[4]arene derivatives with a cone conformation typically possess a higher extraction efficiency than the corresponding monomers due to the size-fitting effect, the complimentary effect and the converging effect of multi-functionality based on their pre-organization, as described in our previous studies [32–45]. In contrast, a calix[4]arene derivative with tetraphosphonic acid at the upper rim exhibited comparable extraction ability to that of its corresponding monomer [29]. The key for obtaining high extraction ability is related to the chelation capacity of the phosphonic acid and phenoxy oxygen atom as functional groups. Propylenephosphonic acid derivative 8 forms a 7-membered chelate ring during the extraction, which is not a preferable chelate form, resulting in low extraction ability. In contrast, methylenephosphonic acid derivative 5 forms a more preferable 5-membered chelate ring, resulting in high extraction ability. Calix[4]arene derivatives with tetraphosphonic acid at the upper rim do not possess phenoxy oxygen atoms for the chelation, and thus exhibit ordinary extraction ability. As support for this explanation, the relationship between pH1/2 values for the monomeric extractants 7 and 9 and the reciprocal number of ionic radii for the rare earth metals is illustrated in Fig. 8. As the chemical formulas and the functionalities of both extractants are nearly the same, their lipophilicities must also be similar. The extraction abilities are, however, completely different. The difference is clearly due to the absence or presence of the phenoxy oxygen atoms, which participate in the chelation of the extractant. The chelation of the functional group and the phenoxy oxygen atom contributes the high extraction ability, as well as the low separation efficiency. In addition to the strong extraction efficiency caused by the phenoxy oxygen atom, the main reason for the poor separation efficiency is the mismatch in the distance between the chelating atoms and the ionic radii of the rare earth metals, especially in the case of the smaller heavier rare earths that are selectively complexed with phosphonic acid, as well as the presence of 1:2 stoichiometry and the absence of a single molecular extraction. In fact, the slope of 7 in Fig. 8 is lower than that of 9, meaning that the separation efficiency of 7 is lower than that of 9. In our previous works, the extraction behavior of acetic acid containing calix[4]arenes with different spacers was investigate [46, 47]. The extraction ability and the separation efficiency were found to be strongly related to the length of the spacer. In this study, the effect of the phenoxy oxygen atom on the extraction behavior is significant, and its presence in the molecular design of calix[4]arene derivatives should be evaluated.

Relationship between pH1/2 values of the monomeric extractants 7 and 9 and the reciprocal number of ionic radii for rare earth metals
Conclusions
A cone conformational p-t-octylcalix[4]arene with four methylenephosphonic acid at the lower rim have been synthesized to investigate the extraction behavior of the nine trivalent rare earth elements. The extractant exhibited significantly higher extraction ability than the corresponding monomer, as well as calix[4]arenes employed in our previous work, containing tetrapropylenephosphonic acid with a longer spacer, tetraphosphonic acid at the upper rim, and tetraacetic acid at the lower rim. Using slope analysis, the stoichiometries of the tetrameric and the monomeric extractants to the rare earth metal ions were determined to be 2:1 and 3:1, respectively. From the proposed extraction equations, the extraction equilibrium constants and separation factors were estimated. The extremely high extraction ability and moderate separation efficiency were attributed to the chelating effect of phosphonic acid and phenoxy oxygen atoms. The effect of the phenoxy oxygen atom on the extraction ability and separation efficiency in calix[4]arene derivatives is significant, and its inclusion in the molecular design of calix[4]arene derivatives should be evaluated.
References
Otu, E.O., Westland, A.D.: Solvent extraction with organophosphonic mono-acidic esters. Solvent Extr. Ion Exch. 8(6), 759–781 (1990)
Peppard, D.F., Driscoll, W.J., Sironen, R.J., McCarry, S.: Nonmonotonic ordering of lanthanides in tributyl phosphate-nitric acid extraction systems. J. Inorg. Nucl. Chem. 4, 326–333 (1957)
Peppard, D.F., Mason, G.W., Maier, J.L., Driscoll, W.J.: Fractional extraction of the lanthanides as their di-alkyl orthophosphates. J. Inorg. Nucl. Chem. 4, 334–343 (1957)
Peppard, D.F., Mason, G.W., Moline, S.W.: The use of dioctyl phosphoric acid extraction in the isolation of carrier-free 90Y, 140La, 144Ce, 143Pr, and 144Pr. J. Inorg. Nucl. Chem. 5, 141–146 (1957)
Peppard, D.F., Mason, G.W., Hucher, I.: Stability constants of certain lanthanide(III) and actinide(III) chloride and nitrate complexes. J. Inorg. Nucl. Chem. 24, 881–888 (1962)
Peppard, D.F., Mason, G.W., Giffin, G.: Extraction of selected trivalent lanthanide and actinide cations by bis(hexoxy-ethyl)phosphoric acid. J. Inorg. Nucl. Chem. 27, 1683–1691 (1965)
Yuan, C., Ye, W., Ma, H., Wang, G., Long, H., Xie, J., Qin, X., Zhou, Y.: Synthesis of acidic phosphates and phosphonates and their structure-reactivity studies on the extraction of neodymium, samarium, ytterbium and yttrium. Sci. Sin. Ser. B 25(1), 7–20 (1982)
Yuan, C., Li, S., Long, H.: Acidic phosphorus esters, synthesis and structure-reactivity studies as ligands. Phosphorous Sulfur Rel. Elem. 18(1–3), 323–326 (1983)
Yuan, C., Yan, J., Feng, H., Long, H., Wu, F., Jin, P.: Extraction chemistry of rare earths by monoalkyl isopropylphosphonates. Sci. Sin. Ser. B 30(7), 681–691 (1987)
Yuan, C., Hu, S.: Correlation analysis in structure and reactivity studies of mono-basic phosphorus esters in rare earth extraction. Sci. Sin. Ser. B 31(2), 137–146 (1988)
Ohto, K., Inoue, K., Goto, M., Nakashio, F., Nagasaki, T., Shinkai, S., Kago, T.: Solvent extraction of trivalent yttrium, holmium, and erbium by novel types of acidic organophosphonates. Bull. Chem. Soc. Jpn. 66(9), 2528–2535 (1993)
Yoshizuka, K., Kosaka, H., Shinohara, T., Ohto, K., Inoue, K.: Structural effect of phosphoric esters having bulky substituents on the extraction of rare earth elements. Bull. Chem. Soc. Jpn. 69(3), 589–596 (1996)
Wakui, Y., Yokoyama, T., Akiba, K.: Solvent extraction of lanthanide (III) with 1,3-benzenedimethylbis(phenylphosphinic acid). Anal. Sci. 14(4), 819–821 (1998)
Goto, M., Matsumoto, S., Uezu, K., Nakashio, F., Yoshizuka, K., Inoue, K.: Development and computational modeling of novel bifunctional organophosphorus extractants for lanthanoid separation. Sep. Sci. Technol. 34(11), 2125–2139 (1999)
Gutsche, C.D. (ed.): Calixarenes Revisited. Royal Society of Chemistry, Cambridge (1996)
Asfari, Z., Boehmer, V., Harrowfield, J.M., Vicens, J. (eds.): Calixarenes 2001. Kluwer, Netherlands (2001)
Izatt, R.M., Pawlak, K., Bradshaw, J.M.: Thermodynamic and kinetic data for macrocycle interactions with cations and anions. Chem. Rev. 91(8), 1721–1785 (1991)
Arnaud-Neu, F.: Solution chemistry of lanthanide macrocyclic complexes. Chem. Soc. Rev. 23(4), 235–241 (1994)
Roundhill, D.M.: Metal complexes of calixarenes. Prog. Inorg. Chem. 43, 533–592 (1995)
Agrawal, Y.K., Kunji, S., Menon, S.K.: Analytical applications of calixarenes. Rev. Anal. Chem. 17(2), 69–139 (1998)
Ludwig, R.: Calixarenes in analytical and separation chemistry. Fresenius J. Anal. Chem. 367(2), 103–128 (2000)
Lumetta, G.J., Rogers, R.D., Gopalan, A.S. (eds.): Calixarenes for separations. In: ACS symposium series. 757, 2nd–17th chapters, Calixarene-cation complexation, 12–236 (2000)
Menon, S.K., Sewani, M.: Chemical modifications of calixarenes and their analytical applications. Rev. Anal. Chem. 25(1), 49–82 (2006)
Sliwa, W., Girek, T.: Calixarene complexes with metal ions. J. Incl. Phenom. Macrocycl. Chem. 66, 15–41 (2010)
Ohto, K.: Review of the extraction of metal cations with calixarene derivatives. Solvent Extr. Res. Dev. Jpn. 17, 1–18 (2010)
Oshima, T., Yamamoto, T., Ohto, K., Goto, M., Nakashio, F., Furusaki, S.: A calixarene-based phosphoric acid extractant for rare earth separation. Solvent Extr. Res. Dev. Jpn. 8, 194–204 (2001)
Jurecka, P., Vojtisek, P., Novotny, K., Rohovec, J., Lukes, I.: Synthesis, characterization and extraction behaviour of calix[4]arene-based phosphonic acids. J. Chem. Soc. Perkin Trans. 2(7), 1370–1377 (2002)
Matulkova, I., Rohovec, J.: Synthesis, characterization and extraction behaviour of calix[4]arene with four propylene phosphonic acid groups on the lower rim. Polyhedron 24, 311–317 (2005)
Ohto, K., Ota, H., Inoue, K.: Solvent extraction of rare earths with a calix[4]arene compound containing phosphonate groups introduced onto upper rim. Solvent Extr. Res. Dev. Jpn. 4, 167–182 (1997)
Ohto, K., Yamasaki, T., Inoue, K.: Extractive separation of rare earth ions by using calix[4]arene with isopropyl hydrogen phosphonate at upper rim. Ars Sep. Acta 4, 96–106 (2007)
Wieser-Jeunesse, C., Matt, D., Yaftian, M.R., Burgard, M., Harrowfield, J.M.: Synthesis and properties of phosphorus-containing calix[4]arenes. C. R. Acad. Sci. Paris 1(8), 479–502 (1998)
Alexandratos, S.D., Natesan, S.: Coordination chemistry of phosphorylated calixarenes and their application to separations science. Ind. Eng. Chem. Res. 39(11), 3998–4010 (2000)
Cherenok, S., Kalchenko, V.: Phosphorus-containing calixarenes. Top Heterocycl. Chem. 20, 229–273 (2009)
Luehr, S., Holz, J., Boerner, A.: The synthesis of chiral phosphorus ligands for use in homogeneous metal catalysis. Chem. Cat. Chem. 3, 1708–1730 (2011)
Bayrakcı, M., Ertul, S., Yılmaz, M.: Solubilizing effect of the p-phosphonate calix[n]arenes towards poorly soluble drug molecules such as nifedipine, niclosamide and furosemide. J. Incl. Phenom. Macrocycl. Chem. 74, 415–423 (2012)
Kalchenko, O., Cherenok, S., Yushchenko, O., Kalchenko, V.: Complexation of calix[4]arenehydroxymethylphosphonic acids with amino acids. Binding constants determination of the complexes by HPLC method. J. Incl. Phenom. Macrocycl. Chem. doi:10.1007/s10847-012-0169-x (2012)
Ohto, K., Matsufuji, T., Yoneyama, T., Tanaka, M., Kawakita, H., Oshima, T.: Preorganized, cone-conformational calix[4]arene possessing four propylenephosphonic acids with high extraction ability and separation efficiency for trivalent rare earth elements. J. Incl. Phenom. Macrocycl. Chem. 71((3,4)), 489–497 (2011)
Ohto, K., Yano, M., Inoue, K., Yamamoto, T., Goto, M., Nakashio, F., Shinkai, S., Nagasaki, T.: Solvent extraction of trivalent rare earth metal ions with carboxylate derivatives of calixarenes. Anal. Sci. 11(6), 893–902 (1995)
Jeanmaire, T., Hervaud, Y.: Synthesis of dialkyl hydroxymethylphosphonates in heterogeneous conditions. Phosphorous Sulfur Rel. Elem. 177(5), 1137–1145 (2002)
Phillion, D.P., Andrew, S.S.: Synthesis and reactivity of diethyl phosphonomethyl triflate. Tetrahedron Lett. 27, 1477–1480 (1986)
Harned, H.S., Owen, B.B.: The physical chemistry of electrolytic solutions, 3rd edn, p. 748. Reinhold Publishing Corporation, New York (1958)
Peppard, D.F., Mason, G.W., Lewey, S.: A tetrad effect in the liquid–liquid extraction ordering of lanthanides(III). J. Inorg. Nucl. Chem. 31, 2271–2272 (1960)
Kolarik, Z., Pankova, H.: Acidic organophosphorus extractants—I extraction of lanthanides by means of dialkyl phosphoric acids—effect of structure and size of alkyl group. J. Inorg. Nucl. Chem. 28, 2325–2333 (1966)
Ohto, K., Murakami, E., Shinohara, T., Shiratsuchi, K., Inoue, K., Iwasaki, M.: Selective extraction of silver (I) over palladium (II) with ketonic derivatives of calixarenes from highly concentrated nitric acid. Anal. Chim. Acta 341(2–3), 275–283 (1997)
Ohto, K., Yamaga, H., Murakami, E., Inoue, K.: Specific extraction behavior of amide derivative of calix[4]arene for silver (I) and gold (III) ions from highly acidic chloride media. Talanta 44(6), 1123–1130 (1997)
Ohto, K., Fujimoto, Y., Inoue, K.: Stepwise extraction of two lead ions with a single molecule of calix[4]arene tetracarboxylic acid. Anal. Chim. Acta 387(1), 61–69 (1999)
Ohto, K., Shiratsuchi, K., Inoue, K., Goto, M., Nakashio, F., Shinkai, S., Nagasaki, T.: Extraction behavior of copper(II) ion by calixarene carboxylates derivatives preorganized by sodium ion. Solvent Extr. Ion Exch. 14(3), 459–478 (1996)
Yoneyama, T., Sadamatsu, H., Kuwata, S., Kawakita, H., Ohto, K.: Allosteric coextraction of sodium and metal ions with calix[4]arene derivatives 2: first numerical evaluation for the allosteric effect on alkali metal extraction with crossed carboxylicacidtypecalix[4]arenes. Talanta 88, 121–128 (2012)
Acknowledgments
This work was financially supported in part by “The Environment Research and Technology Development Fund,” No. K123022, from the Ministry of the Environment, Government of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ohto, K., Takedomi, A., Chetry, A.B. et al. The effect of phenoxy oxygen atoms on extremely high extraction ability and less separation efficiency of trivalent rare earth elements with tetraphosphonic acid derivative of calix[4]arene. J Incl Phenom Macrocycl Chem 77, 363–373 (2013). https://doi.org/10.1007/s10847-012-0255-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-012-0255-0