
Abstract
In molecular modeling the description of the interactions between molecules forms the basis for a correct prediction of macroscopic observables. Here, we derive atomic charges from the implicitly polarized electron density of 11 molecules in the SAMPL6 challenge using the Hirshfeld-I and Minimal Basis Set Iterative Stockholder (MBIS) partitioning method. These atomic charges combined with other parameters in the GAFF force field and different water/octanol models were then used in alchemical free energy calculations to obtain hydration and solvation free energies, which after correction for the polarization cost, result in the blind prediction of the partition coefficient. From the tested partitioning methods and water models the S-MBIS atomic charges with the TIP3P water model presented the smallest deviation from the experiment. Conformational dependence of the free energies and the energetic cost associated with the polarization of the electron density are discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the past 11 years SAMPL challenges have included blind prediction of a variety of different properties such as hydration free energy [1], binding affinity of host–guest systems [2, 3], distribution coefficients [4, 5] and calculations of pKa [6]. They have made an important contribution to the development of new methods and computational tools [7] and increased the accuracy in the prediction of each of these properties. The interest in more efficient and accurate methodologies to predict these properties lies mainly in their pharmaceutical, biochemical and environmental relevance. Indirectly, all these properties are related to the prediction of binding free energies of ligands to proteins.
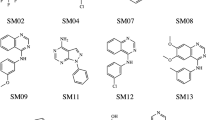
The challenge of SAMPL6 part II consisted in the determination of octanol water partition coefficients of 11 molecules (see Fig. 1) that are similar to fragments of protein kinase inhibitors and are a subset of molecules that were part of the pKa SAMPL6 challenge [6]. The determination of the logarithm of the partition coefficient experimentally [8] and computationally is challenging, and in general there are not many cases in which different computational methodologies are tested blindly (without knowledge of the experimental results).

Set of molecules proposed by the SAMPL6 challenge
From a molecular modeling perspective partition coefficients are important experimental observables used to validate force fields for small drug like molecules. The increased computational power in combination with established free energy calculation methods make their prediction mainly dependent on the description of the force field because phase space is properly sampled in the simulation time. One of the most commonly used methods are molecular dynamics simulations with an explicit solvent description as reported recently by Bannan et al. [9]. In this work the generalized AMBER force field (GAFF) and its corrected dielectric version combined with a new autonomous tool for the creation of the input files (Solvation Toolkit) resulted in logP values with a root mean square error (RMSE) of 1.2 logarithm units compared to the experimental values. Other approaches combined molecular dynamics with an implicit Generalized-Born solvent model for a group of 11,993 molecules evidencing an RSME of 1.14 log units [10].
Better agreement with experiment has been obtained previously with electronic structure calculations and implicit solvation models as SMD, SM8, SM12 and the COSMO variants obtaining a mean absolute error (MAE) of approximately 0.6 log units for a set of 34 organic molecules and 55 fluorinated alcohols and carbohydrates [11]. There is also a wide variety of empirical methods based on atoms and fragments such as KLOGP [12], ALOGP [13], XLOGP [14, 15], which consist of regression models or neural networks that have been trained to reproduce logP values using a large set of experimental data. All electronic structure methods in general have good results partially because they were parameterized with solvation and transfer free energies of neutral solutes in water and different organic solvents [16, 17]. The empirical statistical models based on fragments have the advantage of being fast compared to the other methods, but they have some drawbacks since they tend to overestimate the lipophilicity of large molecules and do not cover the entire chemical space, which creates greater uncertainty in the confidence of the results [10].
Molecular dynamics using explicit solvents make a more complete representation of the systems accounting for conformational changes of the solute and the solvent molecules including specific hydrogen bond interactions. Crucial for the correct prediction of the free energies are the interactions in the system described by the different force fields. In previous challenges, hydration free energies were found to depend significantly on the employed atomic charges in the force field [18]. More recently, we have shown that the electrostatic interactions described by polarized Hirshfeld-I (HI) and Minimal Basis Set Iterative Stockholder (MBIS) atomic charges result in good agreement of hydration free energies in the FreeSolv database and partition coefficients of methylated DNA bases [19, 20].
Based on our previous results, we blindly predict the experimental logP of the 11 molecules by force field based molecular dynamics simulations with the previously proposed atomic charges testing a large number of variables of the simulation protocol such as the initial conformation used in each solvent, the water and octanol solvent model and the total simulation time (especially for octanol). More specifically, we address the capacity of two different methods to derive atomic charges from the polarized molecular electron density employing the theory of atoms in molecules [21]: the S-HI method (Hirshfeld-Iterative atomic charges using the implicit solvent SMD in the calculation of the electron density by electronic structure methods) and the S-MBIS atomic charges (using the alternative MBIS partitioning method). These atomic charges in combination with the other GAFF force field parameters were used to calculate logP values for the 11 molecules of the SAMPL6 challenge with free energy calculations using explicit solvents.
Methods
Based on the provided smiles strings we created conformers with RDKit 2016.09.4 [22] and optimized their structure with the MMFF94s force field keeping only those conformers presenting a root mean square deviations (RMSD) of the heavy atom positions larger than 0.5 Å compared to the most stable one. The obtained geometries were then optimized with the PM7 semiempirical ab initio method with the MOPAC 2016 software. For SM02 also tautomers were studied were the hydrogen atom of the secondary amine group was moved to the closest nitrogen atom on the aromatic ring. This tautomer was more stable at the PM7 level in vacuum, but not in the DFT calculation mentioned below.
Once the conformations obtained by the previous procedure for each molecule were selected, each of the structures was optimized using the ORCA 4.0.0.2 [23] program at the BLYP theory level and the def2-TZVP base set. This was done in vacuo and using the implicit solvent SMD for water and octanol. Besides the test case of SM13 only the conformer with the lowest free energy in each solvent was used as starting structure for the free energy calculations.
Atomic charges
Atomic charges were obtained from the polarized electronic density of the most stable conformer of each of the 11 molecules proposed in the challenge at the BLYP/def2-TZVP level of theory using the SMD implicit solvent [17] for water and octanol. Two methods to partition the electronic density were used: one based on the Hirshfeld-I [24] method and the other based on the Minimal Basis Iterative Stockholder method [25] using the Horton 2.0.0 program [26] as described in previous work [19, 20]. After obtaining the charges, the chemically equivalent atoms by symmetry were averaged using the OpenEye tools (version 2017.2.1).
Free energy and partition coefficients
The free energy of hydration and solvation was obtained by means of alchemical free energy calculations for each of the 11 molecules using standard protocols described in previous works [20, 27] which allow to obtain free energy values with very small uncertainty. The 11 molecules were solvated in approximately 1500 water molecules using the SPC/E [28] and TIP3P [29] water model for the calculation of hydration-free energy. For octanol, approximately 140 molecules were added in a dodecahedron simulation box using the GROMACS simulation package 5.0.4 [30]. Then a short minimization was performed, and the system was equilibrated by 50 ps in a NVT and a NPT ensemble using a time step of 2 fs in combination with stochastic dynamics [31] (\(\tau = 2\) ps) and the Parrinello–Rahman pressure coupling [32] (\(\tau _{p} = 1\) ps) algorithm using the compressibility of water. For octanol, we tested the effect of changing the compressibility (\(7.6 \times 10^{-5}\)\({\text{bar}}^{-1}\)) but the obtained density was the same as the one obtained with the compressibility value of water. The electrostatic interactions were calculated with the Particle–Mesh–Ewald method [33], a cut-off radius of 1.2 nm, a PME-order of 6 and a spacing of 0.1 nm. The van der Waals (vdW) interactions were scaled to zero via a switching function, which switches the potential to zero between 1.0 and 1.2 nm. The neighbor list was updated every ten steps with the verlet cutoff-scheme implemented in GROMACS 5.0.4 [30] and its cut-off was set to 1.2 nm. All bonds were constrained with the LINCS algorithm [34] of order 4 and the isotropic correction to the energy pressure due to missing van der Waals interactions was applied [35].
After the equilibration of the system the free energy of hydration and solvation was calculated using an alchemical path using molecular dynamics simulations where first the electrostatic interactions of the solute with the solvent were turned off through a lambda parameter using the following lambda values [0.00, 0.50, 0.75, 1.00] and subsequently van der Waals interactions were turned off with the following lambda values [00, 0.05, 0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.65, 0.70, 0.75, 0.80, 0.85, 0.90, 0.95, 1.00] using soft core potentials with parameters \(\sigma = 0.3\), \(\alpha = 0.5\) and \(p = 1\). For hydration, a total time of 5 ns was simulated for each lambda value and a time of 15 ns for solvation in octanol. The results of these simulations were analyzed using the alchemical analysis tool [36] with the MBAR method [37] to estimate the values of free energy of hydration and solvation.
In this study, the atomic charges are derived from a polarized molecule due to the implicit solvent model, either water or octanol. These polarization differs between the two solvents and the energy associated with these two processes has to be accounted for in the calculation of the solvation free energies. To calculate the energetic cost, the electronic structure Hamiltonian of the vacuum calculation \({\hat{H}}_{vac}\) was applied on the wave function of each solute polarized by the reaction field of the SMD octanol or water model. The energetic polarization cost \(E_{\mathrm{pol}}\) is the difference between the expectation value of this calculation and the self-consistent-field energy obtained in the calculation in vacuum [20].
This energetic cost was added to the obtained solvation free energies described above and the logP values were calculated at 298 K by the following equation:
where R is the ideal gas constant and T the temperature.
LogP from implicit solvent electronic structure calculations
Additionaly, we also calculated the logP values with electronic structure calculations using the implicit solvent SMD with the ORCA package 4.0.0.2 [23]. The free energy difference between the molecule in vacuum and using the implicit solvent SMD for water or octanol was determined considering the standard state of 1 mol \(\hbox {L}^{-1}\) under the rigid rotor and harmonic oscillator approximation [38].
Results
Conformational analysis of hydration and solvation free energies
To test the effect of using different conformations in the free energy calculations we selected the SM13 molecule because of its large number of rotable single bonds. We identified the three most stable conformers in water and octanol at the BLYP/def2-TZVP level by rotamer generation (RdKit) and geometry optimization. The three conformers differ mainly in the torsional angle between the two phenyl rings and the relative orientation of the methoxy groups (see Fig. 2). For each conformation free energies in each solvent were calculated using the respective S-MBIS atomic charges and the SPC/E water model. All three conformations present the same values within the errors (see Table 1), probably because the flexible character of the molecule and only small variations in the atomic charges of each conformer (see Fig. 2; mean absolute error of the atomic charges between the three conformers does not exceed 0.01 elementary charge units). But, when we corrected the free energies with the polarization energy we observed significant differences between the conformers. For the most stable conformer which has the two phenyl rings aligned on one plane this energy is largest. This is explained by the conjugated π system built by the two planar phenyl rings leading to larger polarizability and its associated energy cost. The different electronic properties of the three conformers are also reflected in the dipole moment of the most stable conformer which is 3.5 D larger in water than in vacuum at this level of theory. The same trends are also observed with the hybrid functional B3LYP which is known to result in smaller dipole moments than BLYP. The electronic response and polarization of the solute is dependent on the dielectric properties of the solvent which results in smaller polarization energy in octanol for all conformers (see Table 1). The polarization energy corrections are in water (octanol) 2–3 kcal/mol (1 kcal/mol) larger for the most stable conformer SM13_A. This conformational dependent polarization energy correction of the logP values increases the value by almost 1 unit for the most stable conformer.

The most stable conformers of SM13 at the BLYP/def2-TZVP level with the SMD model and the S-MBIS atomic charges of the non-hydrogen atoms in water
As will be shown below, our method overestimates logP values and one possible contribution to the error of all molecules with a common substructure as SM13 (e.g. SM02 and SM09) could arise from the overestimated polarization cost of the most stable conformer which is not representative for the not-planar conformation observed in the MD simulations which possess a smaller polarization energy correction and would lead to smaller logP values.
These molecules might, therefore, present one case where the dynamics of the solute and the solvent are required to provide the correct partitioning coefficients, free energies and polarization energy corrections.
Electron density partitioning and water model dependence
Figure 3 shows the calculated logP values for the 11 molecules compared to the experimental references starting from the most stable conformation. There is a significant dependence on the method used to partition the electron density in atomic contributions providing the S-HI and S-MBIS atomic charges. The MBIS atomic charges outperform the ones obtained with the HI partitioning method, which is in agreement with our previous results on hydration free energies for the FreeSolv database. The poor performance of the HI method could be explained by the presence of N-heterocycles in the structure of the 11 molecules, which also presented large deviations in the hydration free energies. The MBIS partitioning method, which does not rely on the electron density of unstable anions for the calculation of the pro-molecular electron density (see [25] for more details), improved the hydration free energy in our previous study and also the logP values as evidenced in this study.

Parity plots of logP values obtained with the S-MBIS and S-HI atomic charges and the SPC/E or TIP3P water model
For both methods a water model dependence is observed although to a lesser extent. The TIP3P model results in better logP values than SPC/E although the latter is known to reproduce properties of liquid water more accurately. One possible explanation is that the GAFF van der Waals parameters are more consistent with the TIP3P water model, which is widely preferred for the simulations using AMBER and GAFF force fields. Since we did not alter these parameters when replacing the atomic charges in the GAFF force field this could explain the slightly better performance of this water model. But, the effect varies between the molecules and is not systematic, which suggest that an electron density based method to derive van der Waals parameters would be desirable to become independent on previously derived non-bonded parameters. Compared to other methods using molecular dynamics simulations and force fields such as CGENF and GAFF participating in this challenge [39] our results present a comparable RMSE when the S-MBIS atomic charges are combined with the TIP3P water model although some molecules present deviations larger than two logP units (see Table 2). Additionally, we also tested the effect of longer simulation times to calculate the octanol solvation free energy for the SM13 molecule using the S-MBIS atomic charges and SPC/E water model. Extending the simulation time per lambda window from five to twenty nanoseconds did not change the free energy by more than 1 kcal/mol.
Functional group corrections of hydration free energies
In our previous study of hydration free energies we were able to assign corrections to the calculated values based on the functional group present in the 613 molecules [20]. These corrections were based on a statistical model assuming independent contribution of the functional groups to the error in the calculated hydration free energy of each molecule. We focused on the most representative functional groups in the FreeSolv database and were able to identify systematic deviations due to their chemical nature.
After the submission of our results, we wanted to test if this correction would improve the obtained logP value, thereby identifying the error contribution from the hydration free energies. In Fig. 4 we show that in all cases the inclusion of the correction improve the logP values suggesting that the prediction of the hydration free energy contributes considerably to the error and its improvement would also have an impact on the quality of the predicted logP values.

Parity plot of logP values calculated with S-MBIS atomic charges and the SPCE water model including the correction per functional group for the hydration free energies derived in Ref. [20] and the experimental value
LogP from SMD solvation model
For the calculation of the atomic charges we had optimized the geometries of all molecules and calculated the vibrational frequencies of all molecules with the SMD solvation model and the BLYP/def2-TZVP method. Based on these data, we also calculated the hydration and octanol solvation free energies under the rigid rotor—harmonic oscillator approximation resulting in the logP values shown in the parity plot of Fig. 5. The small RMSE is comparable to the best predicted values from the COSMOtherm. However, the good performance of the SMD solvation model has to be taken carefully because its parametrization was mainly based on data of octanol solvation free energies and partition coefficients. Therefore, the predictive property for other solvents might vary.

Parity plots of LogP values calculated at the BLYP/def2-TZVP level of theory with the SMD solvation model under the rigid-rotor harmonic oscillator approximation and the experimental reference
Our method does does not rely on experimental free energies and its only input is the polarized electron density which is obtained accurately from low computational cost DFT methods and mostly independent of the solvation model.
Conclusion
The results show that S-MBIS atomic charges derived from the polarized molecular electron densities of the eleven molecules combined with alchemical free energy calculations using explicit solvent (including polarization energy) provide partition coefficients comparable to other small molecule force field. Considering that no parameters have to be adjusted in their derivation and their similar performance to other atomic charge derivation methods we think they provide a promising alternative in the derivation of the next generation small molecule force fields.
Supporting Information
Gromacs input files of all molecules and the calculated hydration and solvation free energies can be downloaded from https://doi.org/10.5281/zenodo.3559197
References
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) J Comput Aided Mol Des 28(3):135
Muddana HS, Gilson MK (2012) J Comput Aided Mol Des 26(5):517
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) J Comput Aided Mol Des 28(4):305
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) J Comput Aided Mol Des 30(11):927
Rustenburg AS, Dancer J, Lin B, Feng JA, Ortwine DF, Mobley DL, Chodera JD (2016) J Comput Aided Mol Des 30(11):945
Bannan CC, Mobley DL, Skillman AG (2018) J Comput Aided Mol Des 32(10):1165
Klimovich PV, Mobley DL (2015) J Comput Aided Mol Des 29(11):1007
Işık M, Levorse D, Mobley DL, Rhodes T, Chodera JD (2019) J Comput Aided Mol Des. https://doi.org/10.1007/s10822-019-00271-3
Bannan CC, Calabró G, Kyu DY, Mobley DL (2016) J Chem Theory Comput 12(8):4015
Daina A, Michielin O, Zoete V (2014) J Chem Inf Model 54(12):3284
Kundi V, Ho J (2019) J Phys Chem B 123(31):6810
Klopman G, Li JY, Wang S, Dimayuga M (1994) J Chem Inf Comput Sci 34(4):752
Ghose AK, Crippen GM (1986) J Comput Chem 7(4):565
Wang R, Fu Y, Lai L (1997) J Chem Inf Comput Sci 37(3):615
Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L (2007) J Chem Inf Model 47(6):2140
Marenich AV, Cramer CJ, Truhlar DG (2013) J Chem Theory Comput 9(1):609
Marenich AV, Cramer CJ, Truhlar DG (2009) J Chem Theory Comput 5(9):2447
Muddana HS, Sapra NV, Fenley AT, Gilson MK (2014) J Comput Aided Mol Des 28(3):277
Lara A, Riquelme M, Vöhringer-Martinez E (2018) J Comput Chem 39(22):1728
Riquelme M, Lara A, Mobley DL, Verstraelen T, Matamala AR, Vöhringer-Martinez E (2018) J Chem Inf Model 58(9):1779
Heidar-Zadeh F, Ayers PW, Verstraelen T, Vinogradov I, Vöhringer-Martinez E, Bultinck P (2018) J Phys Chem A 122(17):4219
Landrum G. Rdkit: Open-source cheminformatics. http://www.rdkit.org
Neese F (2011) Wiley Interdiscip Rev Comput Mol Sci 2(1):73
Bultinck P, Van Alsenoy C, Ayers PW, Carbó-Dorca R (2007) J Chem Phys 126(14):144111
Verstraelen T, Vandenbrande S, Heidar-Zadeh F, Vanduyfhuys L, Van Speybroeck V, Waroquier M, Ayers PW (2016) J Chem Theory Comput 12(8):3894
Verstraelen T, Tecmer P, Heidar-Zadeh F, Boguslawski K, Chan M, Zhao Y, Kim TD, Vandenbrande S, Yang D, González-Espinoza CE, Fias S, Limacher PA, Berrocal D, Malek A, Ayers PW (2015) Horton 2.0.0. Accessed 25 Jan 2018
Ramos Matos G Duarte, Kyu DY, Loeffler HH, Chodera JD, Shirts MR, Mobley DL, Chem J (2017) Eng Data 62(5):1559
Berendsen H, Grigera JR, Straatsma TP (1987) J Phys Chem 91(24):6269
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein M (1983) J Chem Phys 79:926
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) SoftwareX 1–2:19
Van Gunsteren WF, Berendsen HJC (1988) Mol Simul 1(3):173
Parrinello M, Rahman A (1981) J Appl Phys 52(12):7182
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) J Chem Phys 103(19):8577
Hess B, Bekker H, Berendsen H, Fraaije J (1997) J Comput Chem 18(12):1463
Shirts MR, Mobley DL, Chodera JD, Pande VS (2007) J Phys Chem B 111(45):13052
Klimovich PV, Shirts MR, Mobley DL (2015) J Comput Aided Mol Des 29(5):397
Shirts MR, Chodera JD (2008) J Chem Phys 129(12):124105
McQuarrie DA, Simon JD (1997) Physical chemistry: a molecular approach. University Science Books, Sausalito
Procacci P, Guarnieri G (2019) J Comput-Aided Mol Des. https://doi.org/10.1007/s10822-019-00233-9
Acknowledgements
The authors thank financial support by Fondecyt No. 11160193.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Riquelme, M., Vöhringer-Martinez, E. SAMPL6 Octanol–water partition coefficients from alchemical free energy calculations with MBIS atomic charges. J Comput Aided Mol Des 34, 327–334 (2020). https://doi.org/10.1007/s10822-020-00281-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00281-6