
Abstract
A variety of fields would benefit from accurate \(pK_a\) predictions, especially drug design due to the effect a change in ionization state can have on a molecule’s physiochemical properties. Participants in the recent SAMPL6 blind challenge were asked to submit predictions for microscopic and macroscopic \(pK_a\)s of 24 drug like small molecules. We recently built a general model for predicting \(pK_a\)s using a Gaussian process regression trained using physical and chemical features of each ionizable group. Our pipeline takes a molecular graph and uses the OpenEye Toolkits to calculate features describing the removal of a proton. These features are fed into a Scikit-learn Gaussian process to predict microscopic \(pK_a\)s which are then used to analytically determine macroscopic \(pK_a\)s. Our Gaussian process is trained on a set of 2700 macroscopic \(pK_a\)s from monoprotic and select diprotic molecules. Here, we share our results for microscopic and macroscopic predictions in the SAMPL6 challenge. Overall, we ranked in the middle of the pack compared to other participants, but our fairly good agreement with experiment is still promising considering the challenge molecules are chemically diverse and often polyprotic while our training set is predominately monoprotic. Of particular importance to us when building this model was to include an uncertainty estimate based on the chemistry of the molecule that would reflect the likely accuracy of our prediction. Our model reports large uncertainties for the molecules that appear to have chemistry outside our domain of applicability, along with good agreement in quantile–quantile plots, indicating it can predict its own accuracy. The challenge highlighted a variety of means to improve our model, including adding more polyprotic molecules to our training set and more carefully considering what functional groups we do or do not identify as ionizable.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accurate predictions of \(pK_a\) values are of interest in a variety of fields including pharmaceutical research, as absorption, distribution, metabolism, and toxicity can be profoundly affected by changes in ionization state [1, 2]. Other key physiochemical properties, such as lipophilicity, solubility, and permeability are also ionization state dependent [3,4,5,6]. Knowing the likely ionization state of a molecule is also important as preparation for other modeling studies. For example, predictions of distribution coefficients in SAMPL5 demonstrated how dramatically free energy calculations can be affected by a choice in ionization state of a molecule [7, 8]. Calculations of other biomolecular properties, such as protein ligand binding affinities, are similarly affected by choices in ionization state [9].
Because of the importance of \(pK_a\) prediction, and the difficulty of predicting \(pK_a\) values, the SAMPL challenge organizers included a \(pK_a\) prediction component in SAMPL6. Experimental macroscopic \(pK_a\)s were collected for 24 drug like molecules using an established spectrophotometric technique limited to a pH range between 2 and 12. As a part of follow up analysis, a few NMR experiments were performed to determine the microscopic \(pK_a\)s of a select few molecules [10]. Microscopic \(pK_a\)s refer to an equilibrium resulting from removing a specific hydrogen from a molecule and macroscopic \(pK_a\)s describe the process of removing any hydrogen or an overall change in charge state [11, 12]. All experimental data was kept secret from the public to allow participants in the challenge to make blind microscopic and macroscopic predictions for the 24 molecules. Specifically, there were three formats allowed for prediction submission:
-
type I: microscopic \(pK_a\)s,
-
type II: fractional microstate populations as a function of pH, and
-
type III: macroscopic \(pK_a\)s
where microstates refer to a single tautomer of a specific charge state of a molecule. For each type of submission participants were encouraged, but not required, to submit all predictions their model generated for every molecule. SAMPL6 organizers then evaluated predictions based on experimental results for all macroscopic and a select set of microscopic \(pK_a\)s [13]. Details for the challenge including experimental results, all submitted predictions, and an overview analysis are available online (https://github.com/MobleyLab/SAMPL6).
There are many different methods and tools for \(pK_a\) prediction, and a variety can be seen in this special issue on SAMPL6 results. These techniques vary dramatically in scope, computational cost, and accuracy. Historically, a common approach for predicting \(pK_a\) was through linear free energy relationships using empirically determined constants to relate an acid or base to a parent molecule in a known database [14, 15]. A related technique, quantitative structure-property relationships (QSPR), remain popular. These incorporate a variety of molecular and atomistic descriptors [16,17,18]. Some of these techniques have been updated to use more advanced machine learning models such as artificial neural networks [19, 20]. A variety of quantum mechanical descriptors, including partial atomic charges, have also been shown to be promising in QSPR models—due to computational cost, these methods are impractical for a general model and have only been applied to specific types of ionizable groups [6, 21,22,23]. Quantum mechanical calculations from first principles can also be used to calculate \(pK_a\) using a thermodynamic cycle of deprotonation in the gas phase and the hydration free energy of both the protonated and deprotonated molecule [24]. QM calculations are often still limited in accuracy due to the difficulty in calculating hydration free energies of ionized molecules in implicit solvents. The most successful quantum mechanical predictions from first principles also apply an empirical linear correction factor [25, 26].
Here, we introduce a new machine learning model for predicting microscopic and macroscopic \(pK_a\)s. Our goal was to create a universal model which provides predictions that come with accurate estimates of their uncertainty. Most general methods for \(pK_a\) prediction build separate models for each type of ionizable group. Even predictions based on DFT calculations for model input can require specialized models for different ionizable groups [27].
In contrast, we set out to build a single general model which could predict a microscopic \(pK_a\) for any identified ionizable group. We believe that if our features, or input into the machine learning model, are based on the underlying physical and chemical properties responsible for the variation in deprotonation energy, only one model would be necessary. Artificial neural networks have been successful for predicting \(pK_a\), but require substantial training data. We were interested in a machine learning model that could be built from less training data, but did not require an assumption about the shape of the function being fit. Gaussian process regression meets these requirements providing a model based on distributions in feature space. It also automatically incorporates an assessment of uncertainty based on how similar input data is to the training data [28]. Here, we present this new model and our results for the type I and type III components of the SAMPL6 blind challenge.
Computational methods
We built a pipeline to predict the microscopic and macroscopic \(pK_a\)s of a molecule starting from any molecular representation, such as a SMILES string. Our model directly predicts microscopic \(pK_a\)s and then calculates macroscopic \(pK_a\)s. First, we identify all ionizable groups in a molecule and iterate through them to identify all transitions between microstates. In the next step, we convert each microscopic transition into a list of quantitative features. These features are used as input into our Gaussian process regression model which predicts a \(pK_a\) for each micro-transition. The output from these steps is a list of all microscopic \(pK_a\)s for each molecule. Lastly, macroscopic \(pK_a\)s are analytically calculated from a thermodynamic cycle involving all microscopic transitions. Below, each of these steps is described in detail including an overview of how we trained, validated, and tested our model before the SAMPL6 challenge.
A heuristic approach is used to identify aqueous ionizable groups
The first step in processing any molecule, either for training or prediction, is to identify all microscopic transitions. Molecules, in the form of SMILES strings or any common molecular file format, are processed using OpenEye’s OEChem toolkit [29] and one reasonable tautomer of the neutral form of the molecule is chosen. A substructure search is used to identify groups that commonly ionize in water [15] as either acidic:
-
any protonated oxygen atom
-
any protonated aliphatic sulfur atom
-
cyclopentadiene
-
carbon or nitrogen between two strongly electron withdrawing groups
-
arylsulfonamide nitrogens
-
pyrrole-like aromatic nitrogens
-
any atom with a non-dative, formal positive charge and a hydrogen
or basic:
-
aliphatic nitrogen atoms, not a part of amide or sulfonamide groups
-
pyridine-like aromatic nitrogens
-
trivalent aliphatic phosphorous
-
any atom with a non-dative, formal negative charge.
Next, we protonate all basic groups and then iterate through all ionizable groups recursively removing a proton from each in order to identify all micro-transitions. For each transition we store the protonated and deprotonated form of the molecule. OpenEye’s Omega toolkit is then used to generate a low-energy conformation for each form of the molecule [30]. Next, a list of features is calculated to describe the micro-transition between these two forms of the molecule. This feature list will then be used as input for into our Gaussian process model.
Features were chosen to describe physical characteristics
We chose features based on the chemical and physical properties that affect \(pK_a\). The key properties chemists are trained to think about in relation to ionization are the ionizable atom, resonance, inductive effects, steric effects, and solvation. These properties affect the ability of an ionizable group to support a protonated or deprotonated state along with the associated change in formal charge. We also considered the quantum mechanical approach for calculating \(pK_a\) using a thermodynamic cycle involving the gas phase acidity and the solvation free energy of each form of the molecule. Thus, we calculate features to describe the micro-transition using the protonated and deprotonated forms of the molecule in gas and aqueous phase. Using OpenEye Toolkits we calculate a total of ten features for each transition, some for each form of the molecule and some taking differences in properties between the two forms.
-
Difference in enthalpy
-
Mayer Partial Bond order on the bond between hydrogen and the ionizable group
-
AM1-BCC partial charges on multiple atoms, resulting in six charge-related features
-
Difference of solvation free energy
-
Solvent accessible surface area of the deprotonated atom
To begin, we perform a semi-empirical AM1 calculation for each microstate and then extract several properties. The first feature mirrors gas phase acidity by taking the difference in enthalpy between the protonated and deprotonated form of the molecule. For the protonated form of the molecule, the Mayer partial bond order is also calculated for the bond between the ionized atom and the hydrogen to be removed [31,32,33]. AM1-BCC partial charges are calculated for atoms one and two bonds away from the ionized atom [33,34,35]. Previous work established partial charges as a useful feature to predict \(pK_a\) on molecules. These studies used molecule sets with all the same ionizable groups considering the charge on the deprotonated and surrounding atoms [22, 23, 36]. In order to apply our model to all identified ionizable groups, we needed a more general approach. We decided to consider the partial charge of (1) the deprotonated atom and the average partial charge on atoms (2) one bond and (3) two bonds away from this atom, for both the protonated and deprotonated forms of the molecule (Fig. 1). This leads to six features based on the partial atomic charges. Since the AM1 calculations are performed in gas phase, the last two features attempt to capture the affect of solvation on the equilibrium. The difference in solvation free energy of the two forms of the molecule is estimated by a Poisson Boltzmann surface area calculation as implemented in OpenEye’s Szybki toolkit [37, 38]. Lastly, the solvent accessible surface area around the deprotonated atom is determined with OpenEye’s Spicoli Toolkit [39,40,41,42].

We use the partial charge on the deprotonated atom (yellow) and the average partial charge on atoms one bond (purple) and two bonds (orange) away from that atom in both the protonated and deprotonated form of the molecule, making a total of six features involving partial charges
Gaussian process regression provides a simple machine learning model
We built our Gaussian process regression model using the Python package Scikit-learn [43]. A Gaussian process is a nonparametric model which uses a Bayesian approach to sample a posterior distribution of functions [28]. There are two priors set for a Gaussian process, a mean function and a kernel (or covariance) function. As with most Gaussian process models, we set our prior mean function to zero. When initially training and validating the model, we considered a variety of the kernel functions included in Scikit-learn. To choose a kernel and optimize any required parameters, we used a three-fold cross validation method considering the root mean squared error (RMSE), mean error, and correlation coefficient of the training and validating sets (Section "Training, validation, and internal test sets include monoprotic and select diprotic molecules"). The best performing kernel for our purposes was a Matérn kernel—a generalized function between the squared and absolute exponential kernels [28]. This kernel requires a preset parameter \(\nu \) which was optimized to 2.5 for our model. The general form of Matérn kernel is complex including a Bessel function, and with \(\nu =2.5\), our final kernel is the function:
where c and l are trained constants and d is the distance between two feature vectors.
Macroscopic \(pK_a\)s are calculated from microscopic transitions
Our Gaussian process model is trained to predict microscopic \(pK_a\)s which can be used to analytically calculate macroscopic \(pK_a\)s. Most experimentally measured \(pK_a\)s are macroscopic, providing an equilibrium constant for an overall change in total charge. These macroscopic transitions are comprised of multiple microscopic transitions, each of which consists of the removal of one specific hydrogen atom. If \(pK_a\)s, or equilibrium constants, for all microscopic transitions are known, then the macroscopic \(pK_a\) can be analytically calculated using a thermodynamic cycle [6, 15]. For example, for a molecule with two ionizable groups, the macroscopic \(K_a\)’s are:
where a and b are equilibrium constants for the first deprotonation and c and d are for the second deprotonation with the thermodynamic cycle in Fig. 2. Similar, though more complex, cycles can be drawn for polyprotic molecules, allowing us to calculate macroscopic \(pK_a\)s for any provided molecule.

We identify two ionizable groups in SAMPL6 compound SM22; this thermodynamic cycle shows an example of how microscopic transitions with equilibrium constants a, b, c, and d are related to macroscopic equilibrium constants (\(K_{a_1}\) and \(K_{a_2}\))
Training, validation, and internal test sets include monoprotic and select diprotic molecules
Our training set was derived from an extensive experimental \(pK_a\) database Tony Slater curated from four original sources:
-
Dissociation constants of organic bases in aqueous solution, by D.D. Perin (3775 molecules, 8766 \(pK_a\)s) [15];
-
Dissociation constants of organic acids in aqueous solution, by G. Kortum, W. Vogel and K. Andrussow (1063 molecules, 2893 \(pK_a\)s) [44];
-
Dissociation constants of organic bases in aqueous solution, supplement 1972, by D.D. Perin (4275 molecules, 7844 \(pK_a\)s) [45];
-
Ionisation constants of organic acids in aqueous solution, by E.P. Serjeant and Boyd Dempsey (4584 molecules, 10,912 \(pK_a\)s) [46].
This is the same database used for the OpenEye application \(pK_a\) Prospector. To begin, we filtered database entries for experimental measurements which were aqueous (including removing measurements in \(D_2O\)), taken between 20 and 25 °C, and not tagged as very uncertain. This resulted in a set of 9890 molecules with 26,519 experimental measurements. The large number of experimental results compared to number of molecules is not solely due to molecules with multiple \(pK_a\)s; rather, it is primarily due to replicate measurements for certain molecules. In such cases we performed a weighted average, propagating the estimated uncertainties. As we are most interested in biologically relevant ionization, we also removed molecules where the experimental \(pK_a\)s were outside a range of 0–14.
We currently use SciKit-Learn’s out-of-the-box version of Gaussian Process which assumes one expected value for each feature vector, limiting the types of molecules we can use for training. Specifically, polyprotic molecules are not suitable for training input in this approach. Specifically, for polyprotic molecules, there is a feature vector for each microscopic transition, leading to more predicted values than experimental \(pK_a\) values. For example, there are four microscopic transitions for a diprotic molecule but only two macroscopic \(pK_a\) values. Thus, we focused our training set on instances where a microscopic transition can be directly mapped to an experimental macroscopic \(pK_a\). The first set of molecules was perhaps the most obvious—those with only one ionizable group where the microscopic and macroscopic transition are identical. We checked that molecules we identified as having a single ionizable group also only had one experimental measurement. This resulted in 2672 molecules. To expand the diversity of the training set we added a selection of diprotic molecules. For this set we also included molecules where we identified two ionizable groups and two experimental values were reported. Additionally, we required the difference in these two experimental \(pK_a\)s be greater than three log units to assure dominance of a single microstate in estimation of the macroscopic \(pK_a\). There were a total of 286 diprotic molecules in the database that met this requirement. For these molecules, we assumed each macroscopic \(pK_a\) was dominated by only one microscopic transition.
Before training, we removed 10% of these molecules to later serve as an internal test set, resulting in setting aside 243 monoprotic and 29 diprotic molecules, for a total of 301 data points. The training data then consisted of 2186 monoprotic and 257 diprotic molecules. We then split the training data into thirds in order to use a three-fold cross validation method to evaluate the choice of a Gaussian process model and choose a kernel [47]. To evaluate model performance, we considered RMSE, mean error, and correlation coefficients for each training and validation set pair. We judged model performance on training and cross-validation datasets in the context of learning curves for the purposes of model and feature selection. Statistical analysis from our final cross-validation sets and internal test sets can be found in our supporting information. All training data was recombined for our final Gaussian process model used to evaluate our internal test set and make predictions for SAMPL6.
SAMPL6 challenge results
We predict microscopic \(pK_a\) values using a Gaussian process model trained on 2443 mono- and diprotic molecules (2700 data points). Physical and chemical features are calculated for the protonated and deprotonated form of the molecule using OpenEye toolkits (Section "Features were chosen to describe physical characteristics"). Macroscopic \(pK_a\)s are then analytically calculated from a combination of microscopic transitions (Section "Macroscopic pKas are calculated from microscopic transitions"). We used our model to predict microscopic (type I) and macroscopic (type III) \(pK_a\)s for 24 drug like molecules in the SAMPL6 blind challenge [13]. The SAMPL6 submission IDs assigned to our predictions where 6tvf8 (type I) and hytjn (type III). While DLM is a co-organizer of the challenge, none of the authors had any access to the experimental data nor any knowledge of details of the measurements until experimental values and details were publicly released to all participants. The SAMPL6 organizers also asked for optional microstate populations as a function of pH (type II). However, we elected not to participate in that portion of the challenge.
Predictions were matched with experiment to reduce error
In an ideal world we would have a one-to-one match when comparing predicted and experimental results, where each calculated \(pK_a\) has a corresponding experimental value. When SAMPL6 was announced, it included specification for how the experimental \(pK_a\) values would be measured. This included the limitation to perform experiments in a pH range of \(2{-}12\). Following the organizer suggestions, we included predictions for all macroscopic \(pK_a\)s our model predicted including those outside the specified experimental range. Thus, there are many molecules with fewer experimentally determined \(pK_a\)s than we predicted. The organizers considered two matching algorithms and analyzed all challenge submissions with both methods. The first was a closest matching algorithm where each prediction is matched to an experimental value based on the absolute difference between them. If two predictions are paired to the same experimental value then the match with the larger absolute difference is thrown out leading to one less pair used in the analysis. To prevent the loss of data due to multiple pairings, the organizers redid the analysis using a Hungarian matching algorithm instead [48]. In the Hungarian algorithm, the absolute difference is calculated for each pair of prediction to experiment. Then the combination of pairs which reduces the absolute error for that whole molecule is retained. One potential problem in this approach is that it does not account for the natural ordering of \(pK_a\) values, meaning it is possible that the larger of two predictions could be paired with the smaller of two experimental \(pK_a\)s. For example, if a molecule had two experimental \(pK_a\)s 2.15 and 9.58 and a prediction reported values of 0.50 and 1.84, then the final pairs would be (9.58, 0.5) and (2.15, 1.84) as that would result in the smallest absolute error overall. In general, we believe the Hungarian approach is superior as it allows for all possible data to be included, though an ideal algorithm would restrict the order while matching. Fortunately, this reordering did not occur when our predictions were paired with experiment so we used the Hungarian matching to evaluate our performance. All analysis by organizers for all submissions can be found online (https://github.com/MobleyLab/SAMPL6).
Microscopic \(pK_a\) reported for type I predictions
We reported microscopic \(pK_a\)s for all ionizable groups we identified in the SAMPL6 molecules. The first step for any prediction we perform is to identify ionizable groups and then iterate through those groups to find all microscopic transitions. For SAMPL6, all resonance structures of a given microstate were considered to be a single state and assigned a single identification number for the set. We matched each molecular microstate to the proper identification number using a script adapted from the SAMPL6 organizers that identifies identical resonance structures. A full table of microscopic \(pK_a\)s and the script used to find their identification numbers is provided in the supplementary information.
The SAMPL6 organizers initially provided an approximate evaluation of predicted microscopic transitions using macroscopic \(pK_a\)s. Organizers provided experimental macroscopic \(pK_a\)s for all molecules, but experiments for microscopic \(pK_a\)s were only performed for seven molecules [10]. In an attempt to provide feedback on type I predictions, organizers compared experimental data for molecules with only one experimental \(pK_a\) or two \(pK_a\)s with a difference greater than three relative to microscopic predictions. For each molecule, the experimental values were matched to microscopic predictions that resulted in the lowest error, using the same closest and Hungarian pairing algorithms described in "Predictions were matched with experiment to reduce error". These results are available for all submissions on the SAMPL6 GitHub repository [13]. However, here, we have chosen to focus on analysis of directly measured microscopic predictions rather than microscopic predictions with presumed correspondence to macroscopic measurements. While many macroscopic transitions are likely dominated by a single microscopic transition, including those in "Experimental microscopic pKas were measured for two molecules", we cannot know for sure that this is true for every molecule in this set. These results are highly dependent on the particular matching algorithm and no rigorous analysis of the microscopic \(pK_a\) predictions would be possible. Without evidence of which transition is dominant, this pairing does not provide constructive feedback for improving our model, thus we have chosen not to share those results here.
Experimental microscopic \(pK_a\)s were measured for two molecules
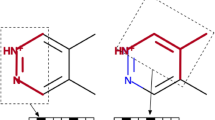
After the macroscopic \(pK_a\) values and all predictions were made public, molecules SM07 and SM14 were analyzed in an NMR experiment to determine microscopic \(pK_a\)s [10]. SM14 has three nitrogens our algorithm identified as ionizable (Fig. 3). The NMR results indicated the two macroscopic \(pK_a\) values were dominated by two different microscopic transitions included in our predictions (Table 1). SM07 is a 4-amino quinazoline derivative with three nitrogens our algorithm identified as ionizable (Fig. 3). The NMR results indicated the macroscopic \(pK_a\) was dominated by a single microscopic transition observed for this molecule. SAMPL6 included five other molecules that are also 4-amino quinazoline derivatives. If we assume all of these molecules have a dominant microscopic transition on the same nitrogen, then we can compare a total of six predicted microscopic \(pK_a\)s with experiment (Table 1). Our predictions for these microscopic transitions have reasonable correlation with the experimental values with an \(R^2\) of \(0.96\pm 0.09\) (Fig. 4). While there appears to be a slight bias with a mean error of \(0.7\pm 0.1\), all predictions were within uncertainty of experiment. Predicted uncertainties are also fairly large, greater than 1.2 for all 4-amino quinazoline derivatives. High uncertainties are expected as our training data only included molecules with one or two ionizable groups and SM14 as well as these 4-amino quinazoline derivatives would not be well represented.

NMR experiments were performed for molecules SM07 and SM14. SM07 has one microscopic transition for the protonation of the top nitrogen (green). SM14 has two microscopic transitions, first the protonation of the middle nitrogen (yellow) and then the left nitrogen (green). For both molecules, we highlight the other nitrogens we identified as ionizable (blue)

Predicted microscopic \(pK_a\)s are compared to experiment for six 4-amino quinazoline derivatives based on NMR experiments on molecule SM07. The shaded region region indicates agreement within \(1\;pK_a\) unit
Commercial models provide a reference for more of our microscopic \(pK_a\) predictions
In an attempt to evaluate a wider range of microscopic \(pK_a\)s, we compared our predictions with some of the top results from the macroscopic analysis. SimulationsPlus’ \(pK_a\) Predictor [20] (submission ID hdiyq) and ACD Lab’s \(pK_a\) GALAS [49] (submission ID 8qph) both performed better than our approach in the macroscopic \(pK_a\) challenge compared to experiment and provided type I predictions [13]. We will refer to these two submissions as S+ and ACD respectively. The SAMPL6 challenge instructions encouraged all participants to submit whatever microscopic \(pK_a\)s their method identified. Each method we are considering for comparison here reported a different number of microscopic \(pK_a\)s. Using our algorithm, we calculated 254 microscopic \(pK_a\)s while, S+ reported 313 and ACD reported 65. In Fig. 5, we compare our results with these two commercial products (Fig. 5 a and b), then we also compare S+ and ACD predictions with each other (Fig. 5c). For all three comparisons, all microscopic transitions predicted by both methods are considered. The two commercial predictions agree reasonably well with one another (RMSE of \(1.9\pm 0.2\)). Thus, comparing our predictions to both allowed for a more informative analysis of our microscopic \(pK_a\) predictions than comparing them to macroscopic experimental data. Our results initially seem less promising than the analysis for SM07 and SM14 discussed above with an RMSD of \(4.7\pm 0.2\) compared to S+ (Fig. 5a) and \(2.9\pm 0.2\) compared to ACD (Fig. 5b). Our model is trained on a limited data set, evident in that it has a smaller dynamic range than either reference method. Therefore, large errors on predictions involving highly charged microstates are to be expected.
If we focus on predictions with uncertainty \(\le 2.0\; pK_a\) units, our microscopic \(pK_a\) predictions are comparable to commercially available tools. Of all the microscopic \(pK_a\) predictions submitted to SAMPL6, only about half the methods included a prediction of uncertainty. Furthermore, ours was the only submission that did not report the same uncertainty value for all predictions. In comparing our results with S+ and ACD we also considered only the points with a predicted uncertainty less than or equal to two \(pK_a\) units (Fig. 5 a and b). Considering this limited set, there is a significant improvement in overall performance with RMSD’s of \(1.8\pm 0.2\) and \(2.1\pm 0.3\) compared to S+ and ACD respectively. These smaller RMSD’s show that when our method predicts a low uncertainty, it agrees with S+ and ACD as well as those two methods agree with each other. Agreement with established commercial models shows promise for our algorithm and reinforces the importance of expanding our limited training set. Additionally, the ability to predict uncertainty demonstrates the reliability of this approach. While predicting uncertainty is a critical feature for model predictions, our model still needs improvement considering S+ and ACD agree on most microscopic \(pK_a\)s and both are more accurate than our method for many macroscopic ionization states.

These plots compare microscopic \(pK_a\) predictions for all combinations of our model, SimulationPlus’ \(pK_a\) predictor and ACD Lab’s \(pK_a\) GALAS. We divide our predictions into two sets: One with predicted uncertainties less than two \(pK_a\) units (orange squares) and greater uncertainties (blue circles). The shaded region indicates an agreement within \(1\;pK_a\) unit. Root mean squared deviation (RMSD) and \(R^2\) are reported for all pairs of points with the same statistics reported in parentheses for low uncertainty points where applicable
Macroscopic \(pK_a\) reported for type III predictions
We reported macroscopic \(pK_a\) values (type III) for all molecules in SAMPL6 (Table 2). These were calculated analytically based on the microscopic \(pK_a\)s determined for each molecule as described in "Macroscopic pKas are calculated from microscopic transitions". Using the Hungarian algorithm described in "Predictions were matched with experiment to reduce error", SAMPL6 organizers compared experimental results with all 34 prediction submissions using RMSE, mean error (ME), and \(R^2\) correlation coefficient. Overall, we saw reasonable agreement between our predictions and experiment (Fig. 6). The SAMPL6 molecules included a variety of polyprotic functional groups that are completely outside the scope of our mono- and diprotic training set. Despite this, over half (18 predictions) fall within one \(pK_a\) unit of experiment. By RMSE and ME we fall within the middle 15 predictions which cannot be easily ranked due to wide confidence intervals, determined using bootstrapping, for most participants. By correlation coefficient (\(R^2\)) our method ranks very low, in the bottom five submissions, but this appears to be due to one rather extreme outlier we discuss in detail below. If we were to remove this one outlier, our ranking would improve significantly with a change in \(R^2\) from \(0.4 \pm 0.1\) to \(0.62 \pm 0.09\) and a shift in RMSE from \(2.2\pm 0.5\) to \(1.7 \pm 0.3\).

This plot shows our macroscopic \(pK_a\) predictions compared to experiment. The shaded region represents agreement within \(1\;pK_a\) unit. The most significant outlier (SM06) is due to an acidic amide we did not identify as ionizable
Molecule SM06 can definitely be considered an outlier, not just due to the large discrepancy between our prediction and experiment, but also due to an ionizable group we did not properly identify (Fig. 7). In this case our predicted value of \(3.94\pm 0.5\) is matched with the experimental value \(11.74 \pm 0.01\), as pointed out in Fig. 6. This molecule contains three ionizable groups: the pyridine nitrogen base, the quinoline nitrogen base and the amide nitrogen either as a base at low pH or as an acid at high pH [50,51,52]. We did not train our model to treat amides as either acids or bases (Section "A heuristic approach is used to identify aqueous ionizable groups"). Our model predicted the transition from + 2 to + 1 to occur at a pH of \(1.77 \pm 2.43\) and to be dominated by deprotonation of the pyridine nitrogen. It predicts + 1 to + 0 transition at \(3.94 \pm 0.54\) dominated by deprotonation of the quinoline nitrogen. In order to improve our model, we consider how similar functional groups are represented in our training set and look to the literature to attempt to determine which microstate dominates at the \(11.74 \pm 0.01\) transition. It seems probable that the deprotonation from + 3 charge to + 2 charge would occur well below pH 2.0, outside the experimental range, and is most likely dominated by the deprotonation of a charged and doubly protonated amide. The next transitions are less immediately obvious, so we look to our training set which contains meta-substituted bromo-pyridines and carboxamide pyridines, both with \(pK_a\)s in the low 3s. It also includes several monoprotic quinoline derivatives with \(pK_a\)s from \(4.8\;\text{to}\;5.5\). One explanation for this large error could be that the carbonyl of the amide group could form an internal hydrogen bond stabilizing the protonated form of the quinoline and increasing its \(pK_a\). While internal hydrogen-bonding may affect some of the features we already include, our model does not directly consider it. Adding a more explicit descriptor to capture such affects may be something we should explore as we improve our model. A more likely explanation for the error is that it is due to the amide nitrogen our model misses. Our logic for not including amides as ionizable sites in our model was because they often have a basic \(pK_a\) value less than 2.0 and an acidic \(pK_a\) value greater than 14.0. However, in this highly conjugated system, that amide nitrogen could be an important contributor to the \(pK_a\) of \(11.74 \pm 0.01\). An analogous system to consider is N-(2-pyrimidyl)benzamide, with its second ionization measured at 11.2 [53], demonstrating that acidic amide nitrogens can have \(pK_a\) values in the appropriate range. Improving our model will likely involve conducting a more thorough investigation of which groups should be considered ionizable.

SM06 provided feedback on our ability to accurate identify ionizable groups as our method only finds two (blue), notably missing the acid amide (red)
We knew going into the SAMPL6 challenge that complex polyprotic molecules would fall outside the domain of applicability for our model, however, other functional groups appear to also be poorly represented. For example, molecule SM20 has only one ionizable group, the acidic imide group (Fig. 8). While our training set includes some similar functional groups, there was not a wide diversity. Specifically, none in the training set had a sulfur one bond away. This is also evident in our prediction \(7.31 \pm 1.84\) where the large uncertainty reflects the lack of similarity between this ionizable group and our training set. Expanding our training data to include polyprotic molecules was already in our plans, but considering more complex mono- or diprotic molecules with overlapping micro-constants could also improve our model.

Our prediction for SM20 was still rather inaccurate, despite it being monoprotic (blue). This is likely due to a lack of representation of imide groups in similar environments in our training set
An important goal in building our model was to be able to predict uncertainties which actually provide some guidance as to expected accuracy and limitations in the training data. Not all commercial products make this a priority; for example, SimulationPlus did not provide uncertainty estimates for any of their predictions. One obvious feature in our data is that the predicted uncertainties are all very large, greater than 1 \(pK_a\) unit for 19 of the matched predictions. Most of the molecules with large uncertainties are polyprotic and include functional groups outside the domain of our training set. Therefore it seems these large uncertainties are a good sign as they seem to correlate with actual error.
Previous SAMPL challenges have included quantile–quantile plots (QQ plots) which provide a more quantitative assessment of a participants reported model uncertainties [7, 54]. QQ plots are based on the concept that actual errors should be drawn from a normal distribution, and well-predicted uncertainties should be able to predict the frequency of deviations of a given size. Thus in QQ plots, y-axis has the fraction of predicted minus experimental values that fall within a given number of uncertainties and the x-axis shows the fraction of a normal distribution within that many standard deviations. The closer the predicted uncertainties compare to a normal distribution the closer they will come to an \(x=y\) line. Thus, the slope of a regression is also often used as a part of the evaluation. We compare two possibilities for model uncertainty in our QQ plot. The first uncertainty approach we consider uses the predicted uncertainties from our Gaussian process model (blue circles in Fig. 9). Another common way to report uncertainty is to assume it is the same for all predictions based on past performance of the approach. For the second set of data, we assumed the uncertainty for each predicted \(pK_a\) was equal to the RMSE for our internal test set, 0.75 (red squares in Fig. 9). A method producing accurate predicted uncertainties should lead to a diagonal line on the resulting plot, with slope of 1; in this case, we find that the uncertainty model using predicted uncertainties from the Gaussian process (slope = 0.87) outperforms the model with a fixed uncertainty (slope = 0.73). This is promising evidence that our model is capable of predicting how its reliability varies with the chemistry being considered, rather than just its overall typical performance.

This QQ plot provides an assessment for predicted uncertainties compared to a normal distribution. Our predicted uncertainties (blue circles) out perform a fixed error of 0.75 taken from our test set RMSE (red squares), as evident by the proximity of the point to the \(x=y\) black line and a slope approaching one
Conclusions
Our Gaussian process model showed promising results in the SAMPL6 challenge, but was limited by the scope of our training set. The chemical space represented in our training set was limited to mono- and diprotic molecules (Section "Training, validation, and internal test sets include monoprotic and select diprotic molecules"). Despite this limitation, we still saw fairly good agreement between our predicted macroscopic \(pK_a\)s and the experimentally measured values and performed competitively compared to other participants. We rank in the top ten by RMSE (\(1.7 \pm 0.3\)) after removing a single obvious outlier (Fig. 6). This outlier, with an acidic amide group, highlighted a potential hole in our limited definition of ionizable groups. Improving our model will require adding groups which are often ionized outside the aqueous \(pK_a\) range, but which can be perturbed to ionize within that range. Our performance in this blind challenge is evidence that a single model trained on physically and chemically relevant features can be competitive with established methods which rely on specialized models for individual functional groups.
The other important step in improving our model will be to augment our training set with additional polyprotic molecules. Currently, the likelihood function in Scikit-learn requires one feature vector for each experimental result. Using this function, we would require a large dataset of experimental microscopic \(pK_a\)s in order to include polyprotic molecules. Generally speaking, it is easier to acquire experimental macroscopic \(pK_a\) data. Thus, a preferred approach would be to define a new likelihood function which would take advantage of the analytical relationship between microscopic and macroscopic \(pK_a\)s and evaluate a set of microscopic predictions with one macroscopic value. We are confident that with this expansion we will have a general model which could predict \(pK_a\) for molecules with any combination of ionizable groups.
Evaluating microscopic \(pK_a\) predictions was limited by the availability of experimental results. For the seven molecules with NMR supported microscopic \(pK_a\)s, our predicted values agreed with experiment within predicted uncertainty. This was a rather limited set of the possible microscopic transitions so we also compared our performance to top-performing commercial products. For microscopic predictions with low predicted uncertainties, our model performs well compared to SimulationPlus and ACD Labs commercial products (Fig. 5). While these results are promising, this portion is a comparison of three predictive models and has not been experimentally verified. A valuable addition to future SAMPL challenges including \(pK_a\) predictions would be to expand experimental measurements to include more microscopic results when available.
We believe predictions are only valuable when they include an accurate assessment of uncertainty, otherwise downstream users have no guidance as to the reliability of such predictions and thus no confidence as to when they can usefully be used and when they should be ignored. These uncertainties are even more valuable if they are determined based on the input molecule, capturing when reliability varies with chemistry. Unfortunately, 10 out of 34 type III submissions in SAMPL6 provided no uncertainties with their predictions. Perhaps requiring such predictions for every submission would improve future challenges and drive progress in this respect. From the beginning, we considered providing an uncertainty evaluation for each prediction an important component of our model. Thus, our ability to determine accurate uncertainty predictions based on input chemistry shows our model’s potential to be a successful predictive method. Previous SAMPL challenges have highlighted the importance and difficulty in accurately assessing model uncertainty for hydration free energies [54] and distribution coefficients [7]. The large error bars for ionizable sites we consider outside our domain of applicability provide evidence our uncertainty estimates are working as desired. QQ plots also support the conclusion that our model is capable of predicting its own uncertainty (Fig. 9).
SAMPL6 was an opportunity to test our Gaussian process model on an external test set and our first completely blind set of predictions. Our new Gaussian process model performed semi-competitively, especially considering its limited training set compared to more established methods which participated. We look forward to incorporating important lessons from this challenge, particularly, expanding our definition of an ionizable group and improving our likelihood function to include polyprotic molecules in our next training set. Overall, SAMPL challenges provide an important service to the community allowing participants to test their predictive models in a blind manner.
Supplementary materials
Included with this article you will find supplementary materials in the form of a PDF with human readable figures and tables and a compressed file with machine readable data and analysis scripts. In the PDF we provide equations for computing macroscopic equilibrium constants \(K_{a_1}\), \(K_{a_2}\), and \(K_{a_3}\) for a triprotic molecule along with the corresponding thermodynamic cycle similar to the one in Fig. 2. Here, we also provide statistics from our cross-validation with our training set and a comparison plot for our internal test set. Also included there is a full list of all microscopic and macroscopic \(pK_a\)s our model predicts for all 24 SAMPL6 molecules. In the electronic materials we include the prediction files we submitted for type I and type III, along with all the analysis scripts we used to generate data and figures provided here. For analysis of all SAMPL6 submissions and details on the experimental data [10] see the GitHub repository provided by challenge organizers (https://github.com/MobleyLab/SAMPL6).
References
Wan H, Ulander J (2006) High-throughput pKa screening and prediction amenable for ADME profiling. Expert Opin Drug Metab Toxicol 2(1):139. https://doi.org/10.1517/17425255.2.1.139
Gleeson MP (2008) Generation of a set of simple, interpretable ADMET rules of thumb. J Med Chem 51(4):817. https://doi.org/10.1021/jm701122q
Manallack DT, Prankerd RJ, Yuriev E, Oprea TI, Chalmers DK (2013) The significance of acid/base properties in drug discovery. Chem Soc Rev 42(2):485. https://doi.org/10.1039/c2cs35348b
Manchester J, Walkup G, Rivin O, You Z (2010) Evaluation of pKa estimation methods on 211 druglike compounds. J Chem Inf Model 50(4):565. https://doi.org/10.1021/ci100019p
Settimo L, Bellman K, Knegtel RMA (2014) Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm Res 31(4):1082. https://doi.org/10.1007/s11095-013-1232-z
Fraczkiewicz R (2013) In silico prediction of ionization. In: Reedijk J (ed) Reference module in chemistry, molecular sciences and chemical engineering. Elsevier, Waltham
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) Blind prediction of cyclohexane–water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des 30(11):1. https://doi.org/10.1007/s10822-016-9954-8
Pickard FC, König G, Tofoleanu F, Lee J, Simmonett AC, Shao Y, Ponder JW, Brooks BR (2016) Blind prediction of distribution in the SAMPL5 challenge with QM based protomer and pKa corrections. J Comput Aided Mol Des 30(11):1. https://doi.org/10.1007/s10822-016-9955-7
Aguilar B, Anandakrishnan R, Ruscio JZ, Onufriev AV (2010) Statistics and physical origins of pK and ionization state changes upon protein-ligand binding. Biophys J 98(5):872. https://doi.org/10.1016/j.bpj.2009.11.016
Işık M, Levorse D, Rustenburg AS, Ndukwe IE, Wang H, Wang X, Reibarkh M, Martin GE, Makarov AA, Mobley DL, Rhodes T, Chodera JD (2018) pka measurements for the sampl6 prediction challenge for a set of kinase inhibitor-like fragments. bioRxiv. https://doi.org/10.1101/368787. https://www.biorxiv.org/content/early/2018/07/13/368787
Darvey IG (1995) The assignment of pKa values to functional groups in amino acids. Biochem Educ 23(2):80. https://doi.org/10.1016/0307-4412(94)00150-N
Bodner GM (1986) Assigning the pKa’s of polyprotic acids. J Chem Educ 63(3):246. https://doi.org/10.1021/ed063p246
Işık M, Rustenburg AS (2018) Michael, Shirts, D.L. Mobley, J.D. Chodera. SAMPL6. https://github.com/MobleyLab/SAMPL6
Exner O (1972) Advances in linear free energy relationships. Springer, Boston. https://doi.org/10.1007/978-1-4615-8660-9_1
Perrin D, Dempsey B, Serjeant E (1981) pKa prediction for organic acids and bases. Chapman and Hall, New York
Geidl S, Svobodová Vařeková R, Bendová V, Petrusek L, Ionescu CM, Jurka Z, Abagyan R, Koča J (2015) How does the methodology of 3D structure preparation influence the quality of pKa prediction? J Chem Inf Model 55(6):1088. https://doi.org/10.1021/ci500758w
Cruciani G, Milletti F, Storchi L, Sforna G, Goracci L (2009) In silico pKa prediction and ADME profiling. Chem Biodivers. 6(11):1812. https://doi.org/10.1002/cbdv.200900153
Katritzky AR, Kuanar M, Slavov S, Hall CD, Karelson M, Kahn I, Dobchev DA (2010) Quantitative correlation of physical and chemical properties with chemical structure: utility for prediction. Chem Rev 110(10):5714. https://doi.org/10.1021/cr900238d
Peterson KL (2000) Reviews in computational chemistry. Wiley, Hoboken
Fraczkiewicz R, Lobell M, Göller AH, Krenz U, Schoenneis R, Clark RD, Hillisch A (2015) Best of both worlds: combining pharma data and state of the art modeling technology to improve in silico pKa prediction. J Chem Inf Model 55(2):389. https://doi.org/10.1021/ci500585w
Citra MJ (1999) Estimating the pKa of phenols, carboxylic acids and alcohols from semi-empirical quantum chemical methods. Chemosphere 38(1):191. https://doi.org/10.1016/S0045-6535(98)00172-6
Vařeková RS, Geidl S, Ionescu CM, Skřehota O, Bouchal T, Sehnal D, Abagyan R, Koča J (2013) Predicting pKa values from EEM atomic charges. J Cheminf 5:18. https://doi.org/10.1186/1758-2946-5-18
Dixon SL, Jurs PC (1993) Estimation of pKa for organic oxyacids using calculated atomic charges. J Comput Chem 14(12):1460. https://doi.org/10.1002/jcc.540141208
Zevatskii YE, Samoilov DV (2011) Modern methods for estimation of ionization constants of organic compounds in solution. Russ J Org Chem 47(10):1445. https://doi.org/10.1134/S1070428011100010
Pracht P, Bauer CA, Grimme S (2017) Automated and efficient quantum chemical determination and energetic ranking of molecular protonation sites. J Comput Chem 38(30):2618. https://doi.org/10.1002/jcc.24922
Bochevarov AD, Harder E, Hughes TF, Greenwood JR, Braden DA, Philipp DM, Rinaldo D, Halls MD, Zhang J, Friesner RA, Jaguar (2013) A high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem 113(18):2110. https://doi.org/10.1002/qua.24481
Bochevarov AD, Watson MA, Greenwood JR, Philipp DM (2016) Multiconformation, density functional theory-based pka prediction in application to large, flexible organic molecules with diverse functional groups. J Chem Theory Comput 12(12):6001. https://doi.org/10.1021/acs.jctc.6b00805
Rasmussen CE, Williams CKI (2006) Gaussian processes for machine learning, adaptive computation and machine learning. MIT Press, Cambridge
OpeneEye Scientific Software, Inc. OEChem Toolkit (2018). http://www.eyesopen.com
Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J Chem Inf Model 50(4):572. https://doi.org/10.1021/ci100031x
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24(3):1083. https://doi.org/10.1016/0040-4020(68)88057-3
Mayer I (2007) Bond order and valence indices: a personal account. J Comput Chem 28(1):204. https://doi.org/10.1002/jcc.20494
OpeneEye Scientific Software, Inc. OEQuacPac Toolkit (2018). http://www.eyesopen.com
Jakalian A, Bush BL, Jack DB, Bayly CI (2000) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem 21(2):132
Jakalian A, Jack DB, Bayly CI (2002) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem 23(16):1623. https://doi.org/10.1002/jcc.10128
Jelfs S, Ertl P, Selzer P (2007) Estimation of pKa for druglike compounds using semiempirical and information-based descriptors. J Chem Inf Model 47(2):450. https://doi.org/10.1021/ci600285n
Nicholls A, Wlodek S, Grant JA (2010) SAMPL2 and continuum modeling. J Comput Aided Mol Des 24(4):293. https://doi.org/10.1007/s10822-010-9334-8
Grant JA, Pickup BT, Nicholls A (2001) A smooth permittivity function for Poisson-Boltzmann solvation methods. J Comput Chem 22(6):608. https://doi.org/10.1002/jcc.1032
Nicholls A (2004) Spicoli: a surface toolkit, dude
Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55(3):379. https://doi.org/10.1016/0022-2836(71)90324-X
Connolly ML (1983) Analytical molecular surface calculation. J Appl Cryst 16(5):548. https://doi.org/10.1107/S0021889883010985
Sharp KA, Nicholls A, Fine RF, Honig B (1991) Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science 252(5002):106. https://doi.org/10.1126/science.2011744
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit-learn: machine learning in python. J Mach Learn Res 12:2825
Kortüm G, Vogel W, Andrussow K (1960) Disssociation constants of organic acids in aqueous solution. Pure Appl Chem 1(2–3):187. https://doi.org/10.1351/pac196001020187
Perrin DD (1972) Dissociation constants of organic bases in aqueous solution: supplement 1972. Butterworths, London
Serjeant P, Dempsey B (1979) Ionisation constants of organic acids in aqueous solution. Pergamon, Oxford
Hastie T, Tibshirani R, Friedman JH (2009) The elements of statistical learning: data mining, inference, and prediction, 2nd edn. Springer, New York
Kuhn HW (2004) The Hungarian method for the assignment problem. Nav Res Logist 52(1):7. https://doi.org/10.1002/nav.20053
Advanced Chemistry Development, Inc. pKa GALAS (2015). www.acdlabs.com
Ripin D, Evans D (2005) pKa table. http://evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf
Goldfarb AR, Mele A, Gutstein N (1955) Basicity of the amide bond. J Am Chem Soc 77(23):6194. https://doi.org/10.1021/ja01628a031
Bordwell FG, Algrim DJ, Harrelson JA (1988) The relative ease of removing a proton, a hydrogen atom, or an electron from carboxamides versus thiocarboxamides. J Am Chem Soc 110(17):5903. https://doi.org/10.1021/ja00225a054
Evans RE (1964) 460. hydropyrimidines. part iii. reduction of amino-pyrimidines. J Chem Soc. https://doi.org/10.1039/JR9640002450
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28(3):135. https://doi.org/10.1007/s10822-014-9718-2
Acknowledgements
DLM and CCB appreciate the financial support from the National Science Foundation (CHE 1352608) and the National Institutes of Health (1R01GM108889-01). CCB was supported financially by OpenEye Scientific Software to build this model during Summer 2017 and is now supported by a fellowship from The Molecular Sciences Software Institute under NSF Grant ACI-1547580. We are thankful for valuable conversations with OpenEye employees, the SAMPL6 organizers, and all challenge participants, and especially to Merck for its contributions to the experimental work in this challenge. AGS would like to thank Paul Hawkins, Christopher Bayly and Robert Tolbert as well as Anthony Nicholls and Matthew Geballe for many insightful discussions of \(pK_a\) and machine learning.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bannan, C.C., Mobley, D.L. & Skillman, A.G. SAMPL6 challenge results from \(pK_a\) predictions based on a general Gaussian process model. J Comput Aided Mol Des 32, 1165–1177 (2018). https://doi.org/10.1007/s10822-018-0169-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0169-z