
Abstract
Aquaporins are a class of ancient channel proteins. The role of aquaporin proteins (AQPs) has been extensively studied in land plants. However, the information on AQPs in seaweeds is unclear. Pyropia yezoensis (Rhodophyta) grows in the intertidal zone and is believed to be a model research organism for seaweeds in the intertidal zone. In this study, we isolated two AQPs from P. yezoensis (PyAQPs). They contained two conserved Asn-Pro-Ala (NPA) motifs and six transmembrane α-helices (H1–H6). Phylogenetic analysis showed that they are classified into two subfamilies, with much greater evolutionary distance with green plants. PyAQP1 belonged to the Glp group, whereas PyAQP2 belonged to a single small group. Subcellular localization showed that PyAQP1 was distributed on the plasma membrane of the cells, and PyAQP2 was mainly distributed in the plasma membrane and cytoplasm of the cells. The two genes displayed differential expression profiles for different abiotic stresses (desiccation, salinity, and temperature) at the mRNA and protein levels. Over-expression of PyAQPs in yeast cells indicated that PyAQPs improved the growth of yeast under osmotic stress. These results provide an insight towards understanding the molecular mechanisms of P. yezoensis responding to stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aquaporins (AQPs) are a class of transmembrane channel proteins involved in the transportation of water and some small molecules across cellular membranes. AQPs belong to the ancient family of major intrinsic proteins (MIPs), which are present in organisms as diverse as archaea, bacteria, plants, insects, and mammals (Chaumont et al. 2001; Johanson et al. 2001; Finn and Cerda 2015). Studies have revealed that AQPs have a wide diversity in both prokaryotic and eukaryotic organisms (Abascal et al. 2014; Finn and Cerda 2015; Verma et al. 2015). The AQP family is divided into two subfamilies. Ordinary aquaporins are originally thought to specifically transport water, whereas glycerol-uptake facilitators, or aquaglyceroporins (GLPs), also convey small organic compounds such as glycerol and urea (Maurel et al. 1994).
The AQPs of higher plants are a large and more diverse superfamily than that found in any other groups. Based on sequence similarity, higher plant AQPs fall into five subfamilies: plasma membrane intrinsic proteins (PIPs), tonoplast intrinsic proteins (TIPs), nodulin 26-like intrinsic proteins (NIPs), small basic intrinsic proteins (SIPs), and the Glp-like intrinsic proteins (GIPs) (Johanson et al. 2001; Gustavsson et al. 2005; Zardoya 2005). So far, GIPs have only been identified in Physcomitrella patens and another closely related moss (Gustavsson et al. 2005). The variation of AQPs in chlorophyte algae is likely to be even higher than that found in land plants. Structural and phylogenetic analyses placed a total of 22 AQPs from nine chlorophyte species into seven subfamilies, in which, PIPs and GLPs were also identified. The additional five subfamilies were unique to green algae (Anderberg et al. 2011). GLPs are inferred to have been derived from a horizontal gene transfer event in bacteria about 1000 mya in algae (Gustavsson et al. 2005).
Although the members of the AQP superfamily can show little sequence similarity, almost all of them share some common structures, such as two highly conserved Asn-Pro-Ala (NPA) motifs and six transmembrane helices (H1–H6) connected by five loops (loop A–loop E). Structurally, functional AQP channels, consisting of a single polypeptide containing a water pore, are assembled in the membrane as tetramers. In each monomer, three loops (A, C, and E) are extracellular, and two loops (B and D) are intracellular. Loops B and E are folded and extend intramembranous hemi-helices containing a NPA constriction motif. These are involved in the formation of the water pore.
Plant AQP genes have been shown to play a central regulatory role in diverse biological processes, including cell elongation and seed germination, and in environmental stresses, which suggest their putative role in plant water relations and cellular water transport (Alexandersson et al. 2005; Boursiac et al. 2005; Maurel 2007). AQPs also facilitate the movement of low-molecular weight uncharged molecules such as glycerol, urea, boric acid, CO2, hydrogen peroxide (H2O2), and ammonia (NH3) through the plant cell membrane and regulate assimilation and transport within the phloem (Uehlein et al. 2003; Maurel 2007).
AQPs are important tuning components that adapt to unfavorable conditions and respond to diverse abiotic stresses such as drought, salinity, and temperature (Maurel et al. 2008; Aroca et al. 2012). Their expression profiles are various, and different AQPs may be either upregulated or downregulated by the same stress (Alexandersson et al. 2005; Guo et al. 2006). Over-expression of AQPs can increase plant sensitivity to salt, dehydration, or cold stress (Katsuhara et al. 2003; Sade et al. 2009; Wang et al. 2011, 2014). AQP response to abiotic factors was not only at the gene transcription level but also at the translation level, especially by posttranslation modification, in which phosphorylation is now established as an important mark of plant PIPs, affecting activity, trafficking, cycling, and partitioning (Verdoucq et al. 2014).
Pyropia yezoensis (Ueda) M.S. Hwang & H.G. Choi (Rhodophyta, Bangiophyceae) grows in intertidal zones. It is an economically important seaweed widely cultivated in China, Japan, and Korea. Its life cycle consists of foliar gametophytes and shell-boring, filamentous sporophytes (conchocelis). The foliar thalli consist of one or two layers of cells, which is different from higher plants. The thallus is periodically exposed to air, where it experiences a variety of potentially stressful environmental conditions, including desiccation, osmotic stress, and high and low temperatures (Davison and Pearson 1996; Xu et al. 2014). These harsh environmental conditions change seaweed cell water content and osmotic potential, further affecting cell physiological metabolism. The differences in cell structures and habitat are supported by genetically controlled physiological mechanisms in the seaweed. In other species, AQPs are established as important molecules for transport of water and solutes; however, there have been few studies of red algae AQPs and their potential role in adaptive responses to the environment. The completion of the transcriptome of P. yezoensis has made it possible to identify and characterize the AQPs in P. yezoensis. The purpose of this research was to identify AQPs of P. yezoensis through transcriptome analysis, understand their evolutionary relationships, and elucidate their expression patterns and putative functions in response to various abiotic stresses.
Material and methods
Algal material and stress treatment
The pure line PY4–7 of Pyropia yezoensis, established by our laboratory, was used for the experiments. Fresh leafy gametophytes were cultured in sterilized seawater with Provasoli’s enriched seawater medium at 10 °C and 50 μmol photons m−2 s−1 with a 12:12 h light/dark cycle before use. The seawater was bubbled continuously with filter-sterilized air and renewed every 3 days.
Different abiotic stress factors were applied, including dehydration, osmotic potential, and temperature. For dehydration stress, the samples were desiccated until the total water content of the thalli reached 80, 50, and 20% of full hydration. For osmotic potential, different amounts of NaCl were added to the seawater until its salinity reached 8, 16, 33, 47, and 60 P.S.U. Sorbitol was also used to regulate osmotic potential. The osmotic stress lasted 4 h. For temperature stress, the gametophytes were held at −8, 0, 10, 15, 20, and 25 °C, respectively for 4 h. Three biological replicates were produced for each condition. All the collected samples were immersed in liquid nitrogen and stored individually at −70 °C for RNA and protein extraction.
Identification of P. yezoensis AQPs and analysis
Putative aquaporin homologous sequences were identified from the PY4–7 line transcriptome data. These sequences were further used as seed sequences to blast against EST, transcriptome (ID PRJNA235353), and genome (ID SRA061934) data of P. yezoensis using the TBLASTN tool. All the sequences were further assembled after removing redundancy using SeqMan software in the DNAStar package. The resulting sequences were analyzed with BLASTx and the ORF prediction was performed using NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). PCR cloning was used to confirm the accuracy of the sequences using the primers (PyAQP1 and PyAQP2) designed based on the assembled PyAQP sequences (Table 1).
The ExPASy translate tool (http://au.exasy.org/tools/dna.html) was used to predict the putative protein sequences for the PyAQPs. NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/) was used to predict the putative phosphorylation sites of the PyAQP amino acid sequences. The transmembrane helices were predicted with the online TopPred program (http://bioweb.pasteur.fr/seqanal/interfaces/toppred.html) as described by Claros and von Heijne (1994), which predicts transmembrane regions using hydrophobicity analysis. Multiple sequence alignments of amino acid sequences were performed using Clustal X with different parameters (Thompson et al. 1997). A phylogenetic tree was constructed using the maximum likelihood method by RA × ML software (Stamatakis 2008).
In silico predicted subcellular localization and experimental validation
WoLF PSORT (http://wolfpsort.org/) and TargetP (http://www.cbs.dtu.dk/ services/TargetP/) were combined to predict subcellular localization of the proteins. In order to validate the prediction of subcellular localization, transient expression analyses were performed using the onion system based on pBI121-35S-GFP binary vector. The coding regions of the PyAQPs without the stop codon were amplified using RT-PCR from the complementary DNA (cDNA) of P. yezoensis and were inserted in-frame at the N-terminal end of GFP to generate PyAQP-GFP fusion constructs. The clone confirmed by sequencing was digested with BamH I and Xba I and then ligated to the linearized PBI121-GFP vector. The recombinant plasmids of 0.5–1 μg were shot into onion epidermal cells using biolistic bombardment. After transformation, the tissues were incubated on MS agar medium in darkness at 23 °C for 20 h. Observation of GFP signals was performed under a Nikon Eclipse 80i fluorescence microscope.
Quantitative real-time PCR
Total RNA was isolated using the RNeasy Plant Mini Kit (OMEGA) in accordance with the manufacturer’s recommended procedure. The cDNA templates were synthesized using a First Strand cDNA Synthesis Kit (Roche, Germany) according to the manufacturer’s instructions. Then, 1 μg total RNA was used to synthesize the first-strand cDNA. Real-time PCR was performed with Light Cycler480 (Roche, Germany). Sequence-specific primers of different PyAQPs (PyAQP3 and PyAQP4), based on their multiple alignments, were designed using Primer 5.0 software (Premier Biosoft International, USA) (Table 1). PyeIF4A and PyTub1 were simultaneously used as internal controls to normalize the amount of messenger RNA (mRNA) in each reaction. The arithmetic square root of their Ct values was as the final Ct of internal control. The 2−ΔΔCt method was used to assess the expression of PyAQPgenes (Livak and Schmittgen 2001). Differences were calculated using one-way analysis of variance (ANOVA) (P < 0.05) in SPSS 19.0 software. The qPCR results shown are the average (±SD) of three biological repeats.
Western blot analysis
Total protein, including membrane protein, was extracted using a Plant Total Protein Extraction Kit (CWBIO, Beijing, China), and Western blot analysis was performed as described previously (Katsuhara et al. 2003). Proteins, about 100 μg sample−1, were fractionated by 10% SDS-PAGE and transferred to a nylon membrane for immunoblotting. Blots were probed with rabbit polyclonal anti-PyAQP antibody to detect PyAQP protein, stripped, and then reprobed by alkaline-phosphatase conjugated anti-rabbit IgG goat antibody or Ponceau staining (amounts of Rubisco) for the loading control. The information about PyAQP1 and PyAQP2 antigen are listed in Table 1. Immunoblot signals were developed using a chemiluminescent substrate (super Signal West Pico, Thermo Scientific, USA) and visualized using MiniChemi 420-K4 (Sagecreation, Beijing, China).
Over-expression of PyAQP in wild yeast
The Saccharomyces cerevisiae strains used in this study were wild-type BY4741 (MATa, his3-Δ1, leu15-Δ, and ura3-Δ). Each yeast strain was maintained in YPD medium at 30 °C. The full-length cDNAs of PyAQP1 and PyAQP2 were ligated into linearized pYC2-NTC vector with restriction sites of Xba I and BamH I using an In-Fusion Cloning Kit (Clonetech). The primers used for amplifying the coding region of PyAQP1 and PyAQP2 are listed in Table 1. Yeast transformants were selected on selective medium lacking uracil (SD-U), and positive clones were further used for the subsequent sensitivity/tolerance testing of pYC2-NTC strains expressing PyAQP1 and PyAQP2. For osmotic stress, the assays were performed in SD-U medium supplemented with either 3.5 M sorbitol or 2.0 M NaCl.
Results
Identification and cloning of the PyAQP1 and PyAQP2 genes
Based on the functional annotation of P. yezoensis NGS transcriptome, three unigenes related to aquaporin were identified. These sequences were further used as queries to search for homologous sequences by BLAST in the transcriptome and genome of P. yezoensis. Further analyses and PCR cloning showed only two genes to have been acquired with full-length coding regions and classical MIPs superfamily conserved domain. They were named as PyAQP1 and PyAQP2 (GenBank accession nos. KT735045 and KT735044).
The cDNA sequences of PyAQP1 and PyAQP2 contained open reading frames of 1002 and 771 bp, respectively, which encode deduced proteins of 333 and 257 amino acids with estimated molecular masses of 34.56 and 25.8 kD and Pis of 6.43 and 6.58, respectively. The genomic sequences of PyAQP1 and PyAQP2 also contained one intron, 200 and 339 bp in length, respectively. Both PyAQPs had a positive GRAVY score, suggesting that they were hydrophobic proteins, which was a necessary characteristic for AQPs. The blast searches indicate that the two proteins share high homology with other MIPs. Domain analysis shows that they contain two Asn-Pro-Ala (NPA) motifs that are considered critical to the substrate selectivity of PyAQP1 and PyAQP2. Six conserved transmembrane α-helices (H1–H6) were also identified in the PyAQP1 and PyAQP2 secondary structures. These were connected by five loops (loops A–E), forming an aqueous transmembrane pore that constitutes the functional core of MIP (Fig. 1).

Alignment of the predicted amino acid sequences for PyAQPs with other GLPs. Identical and similar residues are shaded in colors, respectively. The putative TM domains are shown withTM1-TM5. The two NPA domains, the Ar/R selectivity filter (H2, H5, LE1, LE2) and Froger’s position (P1–P5) are highlighted with line, arrow and star, respectively
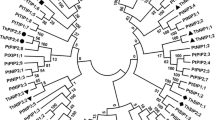
In order to assess the phylogenetic relationships of the PyAQPs, the protein sequences selected as reference sequences were aligned and a phylogenetic tree was constructed using the maximum likelihood method (Fig. 2). The results revealed that the AQPs from the Rhodophyta clustered into three groups (groups I, II, and III), which were not closely associated with the AQPs of land plants. The 22 AQPs of the chlorophyta were clustered into seven groups, two of which were closely associated with the PIP and GIP subfamilies found in land plants. It appeared that the three groups of Rhodophyta were much more similar to the non-plant species. PyAQP1 is prior to cluster with the GLP of bacteria, and this group, comprising both Rhodophyta and bacteria, forms a sister group to the GLPs of animals. This indicates that PyAQP1 belongs to the GLP subfamily of AQPs. PyAQP2 fell into group I, with ChcAQP1 (Chondrus crispus) and GlAQP2 (Gracilaria lemaneiformis), which may be more ancient than group II and group III of the Rhodophyta.

Phylogenetic analysis of PyAQP1, PyAQP2, and other AQPs. An unrooted phylogenetic tree was constructed using the maximum likelihood phylogenetic method in the PHYLIP program
Subcelluar localization of PyAQPs
To verify the sequence-based prediction of the PyAQPs, the subcellular location of the proteins was determined by examination of PyAQP-GFP fusion protein in an onion-epidermal cell transient expression system. As most aquaporins are membrane proteins, the observation was performed after plasmolyzing the onion epidermal cells with 0.6 M sucrose solution. As shown in Fig. 3, for PyAQP1-GFP fusion proteins, the fluorescence signal was mainly distributed along the periphery of the cells; for PyAQP2-GFP fusion protein, the fluorescence signal appeared mainly in the cytoplasm and on the plasma membrane of the cells. As a control, in the cells transformed with the GFP empty vector, the GFP fluorescence signal was distributed throughout the cells.

Subcellular localization of PyAQP1 and PyAQP2 fused with GFP. PyAQP1 and PyAQP2 were transiently expressed in epidermal onion cells and visualized with fluorescence microscopy after 48 h. Fluorescence image of an epidermal cell expressing the p35S-GFP, p35S-PyAQP1-GFP, and p35S-PyAQP2-GFP were visualized under bright field, green light, and blue light, respectively (a is for bright field; b is for green light; c is for blue light). The scale bar corresponds to 40 μm
Expression profiling of PyAQP genes at the mRNA level in different abiotic stress conditions
To better understand the short-term expression patterns of PyAQP in response to abiotic stress, qRT-PCR was used to investigate the expression patterns of PyAQPs under different treatment conditions (salt, desiccation/rehydration, and temperature). PyAQP1 and PyAQP2 were sensitive to water stress to various degrees and also showed different expression patterns. Under desiccation conditions, the expression of PyAQP1 was upregulated significantly and, as the amount of water lost increased, PyAQP1 expression was first upregulated and then downregulated. PyAQP2 was not sensitive to desiccation stress. Only once the desiccation rate reached 80% did PyAQP2 expression decrease significantly. The expression of PyAQP1 and PyAQP2 increased with rehydration after dehydration reached 80% (Fig. 4).

Expression profile analysis of PyAQP1 and PyAQP2 by qPCR under different stresses. The PyeIF4A and PyTub1 genes were used as an internal control for this experiment. Expression fold changes in genes are shown in terms of log 2-fold. The bars show means ± SE from three biological replicates. a Water stress: first bar, control; second bar, dehydration 20%; third bar, dehydration 50%; fourth bar, dehydration 80%; fifth bar, rehydration 15 min after dehydration 80%; sixth bar, rehydration 30 min after dehydration 80%. b Salinity stress: first bar, PSU 8; second bar, PSU 16; third bar, control; fourth bar, PSU 47; fifth bar, PSU 66; sixth bar, low sorbitol (47S); seventh bar, high sorbitol (60S). c Temperature stress: C1: first bar, control; second bar, 0 °C; third bar, -8 °C. C2: first bar, control, second bar, 15 °C; third bar, 20 °C; fourth bar, 24 °C
The sensitivity to osmotic stress of PyAQP is also different. With decreases in salinity below normal, PyAQP1 was significantly upregulated and PyAQP2 expression first decreased and then increased. When salinity was higher than normal, PyAQP1 expression first increased and then decreased; PyAQP2 was repressed as salinity increased. In order to eliminate the effect of osmotic stress from ion concentration changes, different amounts of sorbitol were added to regulate the osmotic pressure. The osmotic pressure of low sorbitol was equal to salinity 47 and high sorbitol to salinity 60. Results revealed that, with the increase in osmotic pressure, the tendency of changes in gene expression were similar to those of increases in salinity. PyAQP1 was upregulated and PyAQP2 was downregulated. It was inferred that, compared to ion concentration, osmotic pressure is the key factor affecting the expression of PyAQP at the mRNA level (Fig. 4).
Under temperature stress, when the temperature was lower than that of normal growth (10 °C), PyAQP1 expression increased significantly, and PyAQP2 expression first decreased and then increased; especially at −8 °C, it was upregulated significantly. When the temperature was higher than 8 °C, both PyAQP1 and PyAQP2 were upregulated (Fig. 4).
Expression profiling of the PyAQP gene at the protein level under different abiotic stress conditions
Desiccation stress induced the expression of PyAQP1 at the protein level, especially at water loss rate of 20%, at which expression level was the highest. After that, PyAQP1 expression slowly decreased. PyAQP2 was not very sensitive to desiccation, and the expression showed the following pattern: decrease-increase-decrease (Fig. 5).

Expression profile analysis of PyAQP1 and PyAQP2 by Western blot after exposure to different stresses. The Rubisco protein was used as an internal control for this experiment. The bars show means ± SE from three biological replicates. a Water stress: first bar, control; second bar, dehydration 20%; third bar, dehydration 50%; fourth bar, dehydration 80%; fifth bar, rehydration 15 min after dehydration 80%; sixth bar, rehydration 30 min after dehydration 80%. b Salinity stress: first bar, PSU 8; second bar, PSU 16; third bar, control; fourth bar, PSU 47; fifth bar, PSU 66; sixth bar, low sorbitol (47S); seventh bar, high sorbitol (60S). c1 Temperature stress: C1: first bar, control; second bar, 0 °C; third bar, -8 °C. C2, first bar, control; second bar, 15 °C; third bar, 20 °C; fourth bar, 24 °C
Under salinity stress, the expression of PyAQP1 increased gradually as salinity decreased. When salinity was increased above normal, PyAQP1 level first increased and then decreased. With the increase in osmotic pressure from sorbitol, PyAQP1 expression was similar to that associated with high salinity (PSU47, 60). The expression of PyAQP2 decreased gradually as salinity decreased. High salinity and high sorbitol induced similar expression patterns, gradual decreases (Fig. 5).
Under temperature stress, PyAQP1 expression increased gradually as the temperature changed from 10 to −8 °C. PyAQP1 was not sensitive to high temperatures and its expression was not increased until 24 °C. PyAQP2 showed similar expression patterns at low temperatures. However, it showed different levels of sensitivity to high temperatures, reaching the highest level at 20 °C and decreasing at 24 °C (Fig. 5).
Over-expression of PyAQPs in wild yeast cells
To further determine the role of PyAQP1 and PyAQP2 for tolerance to salt and osmotic stress, they were ectopically expressed in yeast (S. cerevisiae) using the pYC2/NTC vector. The transformed cell lines expressing PyAQPs and the control lines expressing the empty pYC2/NTC vector were randomly selected to analyze cell growth in the presence of 3.5 M sorbitol and 2.0 M NaCl. As shown in Fig. 6, expression of PyAQP1 and PyAQP2 increased the sensitivity of the yeast to osmotic stress and promoted the growth of the yeast cells on SD-U selective medium. The cells only expressing empty vector and the wild yeast were unable to grow on SD-U medium with osmotic pressure (Fig. 6).

The growth of a yeast cell strain (BY4741) with over-expression of PyAQP genes on SD-U medium with osmolytes. The yeast cell cultures were serially diluted (10-fold) in sterile water and spotted onto plates with different osmotically active media
Discussion
In this work, two AQP genes, PyAQP1 and PyAQp2, were identified from P. yezoensis and bioinformatic analysis showed that they shared the typical structure of MIP proteins, which had a secondary structure containing two highly conserved Asn-Pro-Ala (NPA) constriction motifs and six transmembrane α-helices (H1–H6) connected with five loops (A–D). The NPA motifs were located at the N-terminal end of loop B and loop E. The phylogenetic analyses indicated that PyAQP1 and PyAQP2 were in two different clades. PyAQP1 is much closer to bacterial GLP. PyAQP2, together with AQPs of other red algae, clustered into a sister group independent of the green lineage. GLP was reported to be lost in seed plants, but it was found in mosses and green algae (e.g., Chlamydomonas reinhardtii, Chlorella, Ostreococcus tauri) (Anderberg et al. 2011; Maurel et al. 2015). The AQPs of green algae are more diverse, with PIP and five specific subclasses (MIP A–E) in addition to GIP (Anderberg et al. 2011). For this reason, GIPs are considered to have been acquired early in the algal lineage. In this work, 12 AQPs from five red algal species were divided into three groups. Regarding the divergence of red and green algae, it can be assumed that the AQP family underwent wider variation in the green lineage.
GLPs are permeable to glycerol, urea, and ammonia in addition to water (Agre et al. 2002; Hall et al. 2015). GLPs contain two signature amino acid sequences not found in the classical AQPs, which constricts the transportation of substrates such as glycerol and urea. First, the five (P1–P5) amino acids, especially the Asp (D) (P2) in the second NPA box, were also found in PyAQP1. However, the aromatic residue/arginine (ar/R) showed modifications at S, E, G, and R for PyAQP1 and V, G, F, and R for PyAQP2. These variations may have made the ar/R region wider and more hydrophobic (Hub and de Groot 2008; Ishibashi et al. 2011; Hall et al. 2015).
Abiotic stresses such as salt, drought, and cold affected the water balance of plants and the expression of AQP genes (Aroca et al. 2012; Zhou et al. 2014). This work addressed PyAQP gene expression patterns at the transcription and translation levels under various abiotic stress conditions, including dehydration, salinity, osmotic pressure, and temperature. PyAQPs showed a varied response to different stress treatments. Desiccation upregulated PyAQP1 and downregulated PyAQP2 at the mRNA level. High salinity and sorbitol regulated PyAQP1 and PyAQP2 with the same pattern, consistent with desiccation. Rehydration and low salinity regulated PyAQP with a similar pattern for upregulated PyAQP1 and PyAQP2. These results seemed to indicate that the transcriptional level was an important part of the regulatory process of PyAQPs in response to desiccation, high salinity, and sorbitol. Furthermore, PyAQP1 and PyAQP2 showed differential expression patterns, which indicated that coordinated regulation occurred between the PyAQPs. Both low and high temperatures upregulated the expression of PyAQP1 and PyAQP2. This suggests that low temperature and high temperature had a similar effect on the PyAQPs, which changed the membrane fluidity of P. yezoensis cells and induced PyAQP expression to maintain the cell’s osmotic potential. In plants, the mechanism underlying the differential effects of the upregulation and downregulation of AQPs under abiotic stress conditions is poorly understood. It has been proposed that the downregulation of AQPs might be one way to minimize water loss and that upregulation could be one way to direct water flow into the cell to keep its water potential constant (Suga et al. 2002; Alexandersson et al. 2005; Sade et al. 2009; Venkatesh et al. 2013). Plant AQP genes were expressed in two different stages. During the early stages, plants sensed the water deficits and suppressed the AQP expression to prevent water flow to the outside. After the adaption stage, the expression of AQPs recovered to increase the water inflow (Martinez-Ballesta et al. 2003; Boursiac et al. 2005; Marulanda et al. 2010; Calvo-Polanco et al. 2014; Zhou et al. 2014).
Although the expression pattern of PyAQPs at the translation level is generally consistent with that at the transcription level, the fold of increase or decrease is lower than at the transcription level. Multiple posttranslational modifications of aquaporins played important roles in their regulatory mechanisms targeting aquaporin expression and function, including phosphorylation, methylation, and ubiquitination (Nuhse et al. 2004; di Pietro et al. 2013; Kim et al. 2013; Maurel et al. 2015). It is well established that phosphorylation of aquaporin is important to the regulation of water channel activity (Johansson et al. 2000; Temmei et al. 2005; Mandal et al. 2012; Jang et al. 2014). X-ray structural analysis has suggested that this may result in the opening of the channel and affect water transportation (Nyblom et al. 2009; Qing et al. 2016). Much previous research has experimentally confirmed phosphorylation effects on the aquaporin gating and trafficking. Although different numbers of putative phosphorylation sites are also predicted in the primary structures of PyAQP1 and PyAQP2, this indicated that phosphorylation may also influence the activity of PyAQPs. It is certain that further experiments are needed to validate which sites are necessary for PyAQPs.
Yeast cells have been successfully used for characterization of plant aquaporin. Over-expression AQP in yeast cells can reveal AQP transport ability and substrate selectivity (Maurel et al. 1993; Liu et al. 2003; Bienert et al. 2006; Ma et al. 2006). In this work, PyAQP1 and PyAQP2 were over-expressed in yeast cells and the yeast cell growth was improved obviously under osmotic stress conditions. Because plant aquaporin has a multifunctional channel, it has a wide range of selectivity profiles. These channels transport not only water but also some small solutes, such as H2O2, CO2, NH3, and urea (Gerbeau et al. 1999; Jahn et al. 2004; Loque et al. 2005; Hooijmaijers et al. 2012; Bienert and Chaumont 2014). Glycerol has also been investigated as a solute in animal AQP, in microbial GlpF, and in plant NIP subgroup (nodulin 26-like intrinsic protein). PyAQP1 belongs to the GLP family as indicated by phylogenetic analysis, and PyAQP2 appears to be more ancient than PyAQP1. It can be inferred that the function of PyAQP1 is more similar to that of the GLP family. Filter substrate screening experiments are necessary to confirm the function of these PyAQPs and to understand the roles of PyAQPs in response to abiotic stress.
In conclusion, this study identified and characterized two aquaporin genes of P. yezoensis. Phylogenetic analysis of amino acid sequences divided them into two subgroups: PyAQP1 grouped with GLPs, whereas PyAQP2 localized to a group distant from the plant AQP group. PyAQP1 and PyAQP2 showed coordinated and diverse expression patterns under different abiotic stresses at the mRNA and protein levels. Over-expression PyAQPs improved the yeast cell growth under the osmotic pressure. These results confirmed that the PyAQPs are active in transporting water and thereby maintain osmotic pressure, which adds to our understanding of the molecular stress response mechanisms in P. yezoensis.
References
Abascal F, Irisarri I, Zardoya R (2014) Diversity and evolution of membrane intrinsic proteins. Biochim Biophys Acta 1840:1468–14811
Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S (2002) Aquaporin water channels—from atomic structure to clinical medicine. J Physiol 542:3–16
Alexandersson E, Fraysse L, Sjovall-Larsen S, Gustavsson S, Fellert M, Karlsson M, Johanson U, Kjellbom P (2005) Whole gene family expression and drought stress regulation of aquaporins. Plant Mol Biol 59:469–484
Anderberg HI, Danielson JA, Johanson U (2011) Algal MIPs, high diversity and conserved motifs. BMC Evol Biol 11:110. doi:10.1186/1471-2148-11-110
Aroca R, Porcel R, Ruiz-Lozano JM (2012) Regulation of root water uptake under abiotic stress conditions. J Exp Bot 63:43–57
Bienert GP, Chaumont F (2014) Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta 1840:1596–1604
Bienert GP, Schjoerring JK, Jahn TP (2006) Membrane transport of hydrogen peroxide. Biochim Biophys Acta 1758:994–1003
Boursiac Y, Chen S, Luu DT, Sorieul M, van den Dries N, Maurel C (2005) Early effects of salinity on water transport in Arabidopsis roots. Molecular and cellular features of aquaporin expression. Plant Physiol 139:790–805
Calvo-Polanco M, Sanchez-Romera B, Aroca R (2014) Mild salt stress conditions induce different responses in root hydraulic conductivity of Phaseolus vulgaris over-time. PLoS One 9(3):e90631
Chaumont F, Barrieu F, Wojcik E, Chrispeels MJ, Jung R (2001) Aquaporins constitute a large and highly divergent protein family in maize. Plant Physiol 125:1206–1215
Claros MG, von Heijne G (1994) TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci 10:685-686.
Davison IR, Pearson GA (1996) Stress tolerance in intertidal seaweeds. J Phycol 32:197–211
di Pietro M, Vialaret J, Li GW, Hem S, Prado K, Rossignol M, Maurel C, Santoni V (2013) Coordinated post-translational responses of aquaporins to abiotic and nutritional stimuli in Arabidopsis roots. Mol Cell Proteomics 12:3886–3897
Finn RN, Cerda J (2015) Evolution and functional diversity of aquaporins. Biol Bull 229:6–23
Gerbeau P, Guclu J, Ripoche P, Maurel C (1999) Aquaporin Nt-TIPa can account for the high permeability of tobacco cell vacuolar membrane to small neutral solutes. Plant J 18:577–587
Guo L, Wang ZY, Lin H, Cui WE, Chen J, Liu M, Chen ZL, LJ Q, Gu H (2006) Expression and functional analysis of the rice plasma-membrane intrinsic protein gene family. Cell Res 16:277–286
Gustavsson S, Lebrun AS, Norden K, Chaumont F, Johanson U (2005) A novel plant major intrinsic protein in Physcomitrella patens most similar to bacterial glycerol channels. Plant Physiol 139:287–295
Hall JR, Clow KA, Rise ML, Driedzic WR (2015) Cloning and characterization of aquaglyceroporin genes from rainbow smelt (Osmerus mordax) and transcript expression in response to cold temperature. Comp Biochem Physiol B 187:39–54
Hooijmaijers C, Rhee JY, Kwak KJ, Chung GC, Horie T, Katsuhara M, Kang H (2012) Hydrogen peroxide permeability of plasma membrane aquaporins of Arabidopsis thaliana. J Plant Res 125:147–153
Hub JS, de Groot BL (2008) Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Nat Acad Sci U S A 105:1198–1203
Ishibashi K, Kondo S, Hara S, Morishita Y (2011) The evolutionary aspects of aquaporin family. Am J Physiol Regul Integr Comp Physiol 300(3):R566–R576
Jahn TP, Moller AL, Zeuthen T, Holm LM, Klaerke DA, Mohsin B, Kuhlbrandt W, Schjoerring JK (2004) Aquaporin homologues in plants and mammals transport ammonia. FEBS Lett 574:31–36
Jang HY, Rhee J, Carlson JE, Ahn SJ (2014) The Camelina aquaporin CsPIP2;1 is regulated by phosphorylation at Ser273, but not at Ser277, of the C-terminus and is involved in salt- and drought-stress responses. J Plant Physiol 171:1401–1412
Johanson U, Karlsson M, Johansson I, Gustavsson S, Sjovall S, Fraysse L, Weig AR, Kjellbom P (2001) The complete set of genes encoding major intrinsic proteins in Arabidopsis provides a framework for a new nomenclature for major intrinsic proteins in plants. Plant Physiol 126:1358–1369
Johansson I, Karlsson M, Johanson U, Larsson C, Kjellbom P (2000) The role of aquaporins in cellular and whole plant water balance. Biochim Biophys Acta 1465:324–342
Katsuhara M, Koshio K, Shibasaka M, Hayashi Y, Hayakawa T, Kasamo K (2003) Over-expression of a barley aquaporin increased the shoot/root ratio and raised salt sensitivity in transgenic rice plants. Plant Cell Physiol 44:1378–1383
Kim DY, Scalf M, Smith LM, Vierstra RD (2013) Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell 25:1523–1540
Liu LH, Ludewig U, Gassert B, Frommer WB, von Wiren N (2003) Urea transport by nitrogen-regulated tonoplast intrinsic proteins in Arabidopsis. Plant Physiol 133:1220–1228
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408
Loque D, Ludewig U, Yuan L, von Wiren N (2005) Tonoplast intrinsic proteins AtTIP2;1 and AtTIP2;3 facilitate NH3 transport into the vacuole. Plant Physiol 137:671–680
Ma JF, Tamai K, Yamaji N, Mitani N, Konishi S, Katsuhara M, Ishiguro M, Murata Y, Yano M (2006) A silicon transporter in rice. Nature 440:688–691
Mandal G, Sharma M, Kruse M, Sander-Juelch C, Munro LA, Wang Y, Vilg JV, Tamas MJ, Bhattacharjee H, Wiese M, Mukhopadhyay R (2012) Modulation of Leishmania major aquaglyceroporin activity by a mitogen-activated protein kinase. Mol Microbiol 85:1204–1218
Martinez-Ballesta MC, Aparicio F, Pallas V, Martinez V, Carvajal M (2003) Influence of saline stress on root hydraulic conductance and PIP expression in Arabidopsis. J Plant Physiol 160:689–697
Marulanda A, Azcon R, Chaumont F, Ruiz-Lozano JM, Aroca R (2010) Regulation of plasma membrane aquaporins by inoculation with a Bacillus megaterium strain in maize (Zea mays L.) plants under unstressed and salt-stressed conditions. Planta 232:533–543
Maurel C (2007) Plant aquaporins: novel functions and regulation properties. FEBS Lett 581:2227–2236
Maurel C, Boursiac Y, Luu DT, Santoni V, Shahzad Z, Verdoucq L (2015) Aquaporins in plants. Physiol Rev 95:1321–1358
Maurel C, Reizer J, Schroeder JI, Chrispeels MJ (1993) The vacuolar membrane protein gamma-TIP creates water specific channels in Xenopus oocytes. EMBO J 12:2241–2247
Maurel C, Reizer J, Schroeder JI, Chrispeels MJ, Saier MH Jr (1994) Functional characterization of the Escherichia coli glycerol facilitator, GlpF, in Xenopus oocytes. J Biol Chem 269:11869–11872
Maurel C, Verdoucq L, Luu DT, Santoni V (2008) Plant aquaporins: membrane channels with multiple integrated functions. Annu Rev Plant Biol 59:595–624
Nuhse TS, Stensballe A, Jensen ON, Peck SC (2004) Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell 16:2394–2405
Nyblom M, Frick A, Wang Y, Ekvall M, Hallgren K, Hedfalk K, Neutze R, Tajkhorshid E, Tornroth-Horsefield S (2009) Structural and functional analysis of SoPIP2;1 mutants adds insight into plant aquaporin gating. J Mol Biol 387:653–668
Qing D, Yang Z, Li M, Wong WS, Guo G, Liu S, Guo H, Li N (2016) Quantitative and functional phosphoproteomic analysis reveals that ethylene regulates water transport via the C-terminal phosphorylation of aquaporin PIP2;1 in Arabidopsis. Mol Plant 9:158–174
Sade N, Vinocur BJ, Diber A, Shatil A, Ronen G, Nissan H, Wallach R, Karchi H, Moshelion M (2009) Improving plant stress tolerance and yield production: is the tonoplast aquaporin SlTIP2;2 a key to isohydric to anisohydric conversion? New Phytol 181:651–661
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Systematic Biology 57:758-771
Suga S, Komatsu S, Maeshima M (2002) Aquaporin isoforms responsive to salt and water stresses and phytohormones in radish seedlings. Plant Cell Physiol 43:1229–1237
Temmei Y, Uchida S, Hoshino D, Kanzawa N, Kuwahara M, Sasaki S, Tsuchiya T (2005) Water channel activities of Mimosa pudica plasma membrane intrinsic proteins are regulated by direct interaction and phosphorylation. FEBS Lett 579:4417–4422
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Uehlein N, Lovisolo C, Siefritz F, Kaldenhoff R (2003) The tobacco aquaporin NtAQP1 is a membrane CO2 pore with physiological functions. Nature 425:734–737
Venkatesh J, JW Y, Park SW (2013) Genome-wide analysis and expression profiling of the Solanum tuberosum aquaporins. Plant Physiol Biochem 73:392–404
Verdoucq L, Rodrigues O, Martiniere A, Luu DT, Maurel C (2014) Plant aquaporins on the move: reversible phosphorylation, lateral motion and cycling. Curr Opin Plant Biol 22:101–107
Verma RK, Prabh ND, Sankararamakrishnan R (2015) Intra-helical salt-bridge and helix destabilizing residues within the same helical turn: role of functionally important loop E half-helix in channel regulation of major intrinsic proteins. Biochim Biophys Acta 1848:1436–1449
Wang LL, Chen AP, Zhong NQ, Liu N, XM W, Wang F, Yang CL, Romero MF, Xia GX (2014) The Thellungiella salsuginea tonoplast aquaporin TsTIP1;2 functions in protection against multiple abiotic stresses. Plant Cell Physiol 55:148–161
Wang X, Li Y, Ji W, Bai X, Cai H, Zhu D, Sun XL, Chen LJ, Zhu YM (2011) A novel Glycine soja tonoplast intrinsic protein gene responds to abiotic stress and depresses salt and dehydration tolerance in transgenic Arabidopsis thaliana. J Plant Physiol 168:1241–1248
Xu Y, Chen C, Ji D, Hang N, Xie C (2014) Proteomic profile analysis of Pyropia haitanensis in response to high-temperature stress. J Appl Phycol 26:607–618
Zardoya R (2005) Phylogeny and evolution of the major intrinsic protein family. Biol Cell 97:397–414
Zhou L, Wang C, Liu R, Han Q, Vandeleur RK, Du J, Tyerman S, Shou H (2014) Constitutive overexpression of soybean plasma membrane intrinsic protein GmPIP1;6 confers salt tolerance. BMC Plant Biol 14:181
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 31672641, Grant No. 31372517) and National High Technology Research and Development Program of China (Grant No. 2012AA10A401).
Author information
Authors and Affiliations
Corresponding author
Additional information
Junqing Yang and Na Li contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kong, F., Yang, J., Li, N. et al. Identification and characterization of PyAQPs from Pyropia yezoensis, which are involved in tolerance to abiotic stress. J Appl Phycol 29, 1695–1706 (2017). https://doi.org/10.1007/s10811-016-1042-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-016-1042-x