
Abstract
Lysophosphatidylinositol (LysoPI), an endogenous ligand for G protein-coupled receptor (GPR) 55, has been known to show various functions in several tissues and cells; however, its roles in the central nervous system (CNS) are not well known. In particular, the detailed effects of LysoPI on microglial inflammatory responses remain unknown. Microglia is the immune cell that has important functions in maintaining immune homeostasis of the CNS. In this study, we explored the effects of LysoPI on inflammatory responses using the mouse microglial cell line BV-2, which was stimulated with lipopolysaccharide (LPS), and some results were confirmed also in rat primary microglia. LysoPI was found to reduce LPS-induced nitric oxide (NO) production and inducible NO synthase protein expression without affecting cell viability in BV-2 cells. LysoPI also suppressed intracellular generation of reactive oxygen species both in BV-2 cells and primary microglia and cytokine release in BV-2 cells. In addition, LysoPI treatment decreased phagocytic activity of LPS-stimulated BV-2 cells and primary microglia. The GPR55 antagonist CID16020046 completely inhibited LysoPI-induced downregulation of phagocytosis in BV-2 microglia, but did not affect the LysoPI-induced decrease in NO production. Our results suggest that LysoPI suppresses microglial phagocytosis via a GPR55-dependent pathway and NO production via a GPR55-independent pathway. LysoPI may contribute to neuroprotection in pathological conditions such as brain injury or neurodegenerative diseases, through its suppressive role in the microglial inflammatory response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Microglia are immune cells of the central nervous system (CNS) that survey the environment in a quiescent state by extending and protruding their processes. By detecting abnormalities, such as insults or inflammation in the CNS, microglia are activated to maintain CNS homeostasis by phagocytosing dead cells or pathogens and releasing cytokines [1,2,3,4]. However, aberrantly activated microglia damage neurons by producing excess amounts of nitric oxide (NO), reactive oxygen species (ROS), and cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, eventually causing neuronal dropout by phagocytosing injured neurons [1, 2, 5]. Therefore, adequate control of microglial function in neuroinflammation is essential.
Lysophospholipids (LPLs) are phospholipids that have lost a fatty acid from the sn-1 or sn-2 position by phospholipase A1 or A2, respectively [6, 7], and are classified according to their polar group in the structure. Recently, increasing evidence has indicated that LPLs have several different functions in various types of cells via each receptor [8, 9]. In addition, it has been reported that some LPLs have effects on inflammation and immunity. For example, some types of LPLs, including lysophosphatidic acid, sphingosine-1-phosphate, and lysophosphatidylcholine, produce proinflammatory cytokines such as IL-1β and IL-6 [10,11,12]; however, other types of LPLs, including lysophosphatidylserine and lysophosphatidylethanolamine, exert an anti-inflammatory effect [13, 14]. As for lysophosphatidylinositol (LysoPI), some reports showed that it enhanced extracellular signal-regulated kinase (ERK) or nuclear factor of activated T cell activity, leading to the induction of inflammatory responses [15, 16]. Furthermore, some report pro-inflammatory effects and others report anti-inflammatory effects of the same LPLs [7, 17,18,19]. These observations imply the complexity of the effect of LPLs on inflammation and immunity, which may arise from the difference in the position, unsaturation degree, and/or length of the acyl chain. Thus, further studies are needed to reveal the precise effects of LPLs in each type of tissue and cells.
LysoPI is an endogenous ligand for G protein coupled receptor (GPR) 55, although other candidate receptors for LysoPI have also been suggested [7]. Celorrio et al. [20] found that the expression of GPR55 in the striatum was downregulated in a mouse model of Parkinson’s disease, and activation of GPR55 by agonists relieved haloperidol-induced catalepsy in the mouse model. Thus, it has been suggested that GPR55 can be a therapeutic target for the treatment of neurodegenerative disorders including Parkinson’s disease [7, 20, 21]. Additionally, another study showed that the rat brain contains a significant amount of LysoPI [22]. However, the functions of LysoPI and its receptor GPR55 in the CNS are not fully understood. As for the microglia, Pietr et al. [23] demonstrated that GPR55 was highly expressed by mouse primary microglia and BV-2 cells and its expression was inversely affected by LPS or IFN-γ stimulation, respectively. They also showed that the pre-stimulation with IFN-γ, which enhanced GPR55 expression, boosted LysoPI-induced phosphorylation of ERK in BV-2 microglia, but did not investigate the effects of LysoPI on inflammatory responses by activated microglia. Moreover, Kallendrusch et al. [24] found that LysoPI exerts a neuroprotective effect against N-methyl-D-aspartate-mediated neuronal excitotoxicity depending on GPR55 in rat organotypic hippocampal slice cultures. However, the question of whether LysoPI affects the immune/inflammatory functions of the microglia and the detailed mechanisms by which LysoPI exerts neuroprotection from the activated microglia remain unclear.
In this study, we investigated the effects of LysoPI on inflammatory responses in the microglia that were stimulated experimentally by LPS to reveal whether LysoPI exerts a neuroprotective role via microglia. Furthermore, if LysoPI exerts neuroprotective functions by suppressing the microglial inflammatory responses, we also explored how LysoPI suppresses the microglial inflammatory responses. LysoPI treatment suppressed LPS-induced neuroinflammatory responses such as NO, ROS, and cytokine production and phagocytic activity by microglia. These results suggest that the suppressive effect of LysoPI on microglial inflammatory responses may contribute to neuroprotection.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Lipopolysaccharide (LPS; derived from E. coli 0127:B8), anti-β-actin antibody, 2′,7′-dichlorofluorescin diacetate (H2DCFDA), and protease inhibitor cocktail were bought from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS), and horse serum were bought from Gibco BRL (Grand Island, NY, USA). l-α-Lysophosphatidylinositol sodium salt (LysoPI; derived from bovine liver, mainly composed of C18:0) was obtained from Avanti Polar Lipids (Alabaster, Alabama, USA). LysoPI ammonium salt (derived from porcine liver, mainly composed of C18:0) was purchased from Olbracht Serdary Research Laboratories (Toronto, Ontario, Canada). O-1602 (5-Methyl-4-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-1,3-benzenediol) and CID16020046 (4-[4,6-dihydro-4-(3-hydroxyphenyl)-3-(4-methylphenyl)-6-oxopyrrolo[3,4-c]pyrazol-5(1H)-yl]-benzoic acid) were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA). 2,3-Diaminonaphthalene (DAN) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Dojindo (Kumamoto, Japan). Dr. H. Ihara of Osaka Prefecture University Graduate School of Science kindly provided antibody against iNOS for us. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) antibody was obtained from Bio-Rad (Hercules, CA, USA). Immobilon™ Western Chemiluminescent HRP substrate was bought from Millipore Co. (Billerica, MA, USA).
Cell Cultures
This study was approved by the ethics committees for animal experimentation at Osaka Prefecture University (Permit No.: 30-33) and carried out according to the Guideline for Animal Experimentation at Osaka Prefecture University. Rat primary microglia were prepared from the whole brains of 1-day-old newborn Wistar rats as described previously [25] with slight modification. In brief, the brains were triturated with DMEM using a Pasteur pipette, and dissociated by 0.25% trypsin in Ca2+ and Mg2+-free phosphate-buffered saline containing 5.5 mM glucose for 15 min at 37 °C with gentle shaking. To inactivate trypsin, an equal volume of horse serum containing 0.1 mg/mL DNase I was added to the medium, and then the tissues were centrifuged at 350×g for 5 min. The tissue pellets were suspended with DMEM supplemented with 10% FBS, 100 mg/L streptomycin, and 5 × 104 units/L penicillin. The cell suspension was plated onto polyethyleneimine-coated plastic bottles. The cells were maintained at 37 °C in a 95% air/5% CO2 condition, and the medium was replaced once a week. On 14 days in vitro, microglia were collected from mixed glial cell culture by shaking the plastic bottles at 100 rpm for 1 h, and seeded on 96-well or 24-well plates for suspension culture at a density of 4 × 105 cells/mL. After 1 h, to remove non-adherent cells, the medium was changed and the remaining microglia were cultured for 1 day in 10% FBS-containing DMEM. Then, cells were stimulated with LPS with or without LysoPI. In our culture, more than 95% of the cells were positive to anti-Iba-1 antibody (rabbit polyclonal; Bio-Rad (Hercules, CA, USA)).
The Laboratory of Molecular Pharmacology at Kanazawa University Graduate School kindly provided mouse microglial cell line BV-2 cells for us. BV-2 cells were cultured in DMEM with 10% FBS and antibiotics (streptomycin (100 mg/L)/penicillin (50,000 units/L)) at 37 °C in a 95% air/5% CO2 condition using a dish or plate for the suspension culture (Sumilon, Sumitomo Bakelite Co., Tokyo, Japan). Cells were passaged once a week.
Although it has been reported that some different characteristics exist between BV-2 cells and primary microglia [26, 27], BV-2 cells are widely used as a substitute for primary microglia in a great number of studies. Thus, we used both BV-2 cells and primary microglia to confirm that primary microglial cells can be replaced by BV-2 cells to assess the effects of LysoPI on microglial inflammatory responses.
Nitrite Assay
We evaluated NO released by BV-2 microglia as the level of nitrite (NO2−), a relatively stable metabolite of NO. The level of nitrite was measured as previously described [25]. BV-2 cells were seeded on a 96-well plate. After 24 h, cells were stimulated with 100 ng/mL LPS with or without LysoPI in serum-free DMEM for 24 h. The reason why we stimulated cells under serum-free conditions is that serum proteins such as albumin prevent LPLs from functioning by binding to them [28]. Amounts of nitrite accumulated in the medium were determined fluorometrically using DAN reagent with a microplate reader (ARVO 1420 Multilabel counter; Wallac, PerkinElmer Co.) at an excitation wavelength of 355 nm and emission wavelength of 460 nm.
Cell Viability Assay
Cell viability was assessed as the total mitochondrial activity using MTT assay as described previously [29]. Cells were seeded on a 96-well plate. After 24 h, cells were stimulated with LPS in the presence or absence of LysoPI in serum-free DMEM for 24 h, and then incubated with MTT solution for 1 h at 37 °C, and the generated formazan was dissolved in DMSO. The absorbance was measured at 585 nm with a microplate reader (ARVO 1420 Multilabel counter).
Immunoblotting
We quantified iNOS protein expression by immunoblotting analysis. BV-2 cells were plated in a non-coating 35-mm dish for suspension culture (Sumilon). After 24 h, cells were treated with LPS with or without LysoPI in serum-free DMEM for 24 h. After preparation of cell lysates, samples were electrophoresed and immunoblotted as described previously [29]. Immunoblotting was conducted with primary antibodies against iNOS (1:1000) or β-actin (1:100,000) and secondary antibody, HRP-conjugated anti-mouse IgG antibody (1:10,000). Proteins that reacted with these antibodies were detected by Immobilon™ Western Chemiluminescent HRP Substrate. Detection bands were analyzed by Lumino Image Analyzer (LAS-4000, Fujifilm, Tokyo, Japan). Protein concentrations were measured by Bradford’s method [30] using Coomassie brilliant blue color solution (Nacalai Tesque, Kyoto, Japan) with bovine serum albumin as a standard, in accordance with the manufacturer’s protocol.
Intracellular ROS Assay
ROS generated in BV-2 cells and primary microglia was measured with H2DCFDA, a cell permeable fluorescent dye. Cells were seeded on a 96-well plate. After 24 h, cells were stimulated with LPS in the presence or absence of LysoPI in serum-free DMEM for 6 h, and then incubated with 20 μM H2DCFDA in serum-free DMEM at 37 °C for 30 min. After that, cells were rinsed with Hepes-buffered saline (HBS) containing Ca2+ and Mg2+ twice in a dark room. Finally, HBS (100 μL/well) was added to the plate and intracellular ROS levels were assessed using a microplate reader (ARVO 1420 Multilabel counter) at an excitation wavelength of 485 nm and emission wavelength of 535 nm, as the fluorescent intensity of dichlorofluorescein (DCF).
Phagocytosis Assay
The phagocytic activity of BV-2 cells and primary microglia was evaluated as uptake of carboxylate-modified fluorescent microbeads (FluoSpheres®; orange fluorescence, 1 μm diameter). Cells were seeded on a non-coating 24-well plate for suspension culture (Sumilon). After 24 h, cells were stimulated with LPS in the presence or absence of LysoPI in serum-free or 0.1% FBS-containing DMEM for 24 h. Then, the cells were incubated with microbeads (final concentration 0.013%) for 1 h at 37 °C. After incubation, the cells were washed with DMEM three to four times and observed under a fluorescent microscope (Olympus, IX70). Twenty cells were randomly collected from one well to determine cellular fluorescence intensity using Image J software, and mean fluorescence intensity was calculated.
Cytokine Assay
For measurement of cytokine production, BV-2 cells were seeded on a non-coating 35-mm dish for suspension culture. After 24 h, cells were treated with LPS with or without LysoPI in serum-free DMEM for 24 h. Then, culture supernatants were collected in microtubes and added to a 96-well assay plate. Concentrations of cytokines in supernatants were determined using Bio-Plex MAGPIX (Bio-Rad, USA) according to the manufacturer’s protocol.
Statistical Analysis
Data are indicated as the mean ± standard deviation. For statistical analysis of the data, a Tukey multiple-comparison procedure following one-way analysis of variance (ANOVA) was employed. The results were considered statistically significant when p < 0.05.
RESULTS
LysoPI Reduced the NO Production by LPS-Stimulated BV-2 Microglia
Cultured BV-2 cells were stimulated with 100 ng/mL LPS in the presence of several concentrations of LysoPI for 24 h. LysoPI significantly attenuated NO production by LPS-stimulated BV-2 at 20 μM and 30 μM to the control level (Fig. 1a). LysoPI significantly decreased cell viability at 30 μM (Fig. 1b), but did not affect it at concentrations of up to 20 μM. For further experiments, we used 20 μM LysoPI, because this LysoPI concentration was most effective without affecting cell viability. We measured the expression of iNOS protein by immunoblotting analysis to investigate the mechanism by which LysoPI decreased NO production by LPS-stimulated BV-2 microglia. BV-2 cells were treated with LPS in the presence or absence of LysoPI (20 μM) for 24 h, and then the iNOS protein expression was measured. LysoPI significantly reduced LPS-stimulated expression of iNOS protein in BV-2 cells as well as NO production (Fig. 1c), although the effect was not complete (p < 0.01, control vs LysoPI + LPS). These results suggest that LysoPI reduced LPS-simulated NO production by a decrease in expression of iNOS protein.

Suppressive effect of LysoPI on NO production in LPS-stimulated microglia. BV-2 cells were incubated with 100 ng/mL LPS together with various concentrations (10, 20, 30 μM) of LysoPI for 24 h. Nitrite concentration in the medium was determined by fluorescent assay with DAN (a). Cell viability was assessed by MTT assay (b). BV-2 cells were stimulated with 100 ng/mL LPS with or without 20 μM LysoPI for 24 h to detect expression of iNOS protein by immunoblotting (c). The photograph shows typical bands for iNOS and β-actin protein. The graph shows the iNOS/β-actin ratio of the density of detection bands. Data are shown as relative value to LPS in immunoblotting analysis. Data are mean ± S.D. of 3–9 samples. **p < 0.01, compared to non-treated control, ##p < 0.01, compared to LPS only (LysoPI 0 μM); Tukey multiple-comparison procedure following one-way ANOVA [a F (7, 40) = 71.8, p < 0.01; b F (7, 40) = 9.64, p < 0.01; c F (3, 28) = 42.3, p < 0.01].
LysoPI Reduced the Intracellular ROS Generation by LPS-Stimulated Microglia
To reveal whether LysoPI affects the production of other pro-inflammatory factors, we measured intracellular ROS generation in LPS-stimulated BV-2 cells. Cultured BV-2 cells were treated with LPS with or without LysoPI (20 μM) for 6 h, and then we determined the intracellular generation of ROS by DCF assay. LysoPI significantly reduced the intracellular ROS generation in BV-2 cells without LPS (Fig. 2a). The intracellular ROS generation was significantly increased by LPS. The increased generation of ROS by LPS was also significantly reduced by LysoPI to the control level. Also, in primary microglia, we examined the effect of LysoPI on intracellular ROS generation to confirm the results in BV-2 cells. The cells were incubated with LPS in the absence or presence of 10 μM LysoPI for 6 h. LysoPI treatment did not affect basal ROS generation, but a significant decrease in ROS generation by LysoPI was observed in LPS-stimulated primary microglia, similarly to BV-2 cells (Fig. 2b).

Effect of LysoPI on LPS-induced generation of ROS in microglia. BV-2 cells (a) and rat primary microglial cells (b) were incubated with LPS in the presence or absence of 10 (b) or 20 μM (a) LysoPI for 6 h, and then intracellular ROS generation, was detected by fluorescent assay with H2DCFDA. Data are shown as relative value to control. Data are the mean ± S.D. of 8–13 (a) or 4 (b) samples. **p < 0.01, compared to control; ##p < 0.01, compared to LPS; Tukey multiple comparison procedure following one-way ANOVA [a F (3, 38) = 16.7, p < 0.01; b F (3, 12) = 11.2, p < 0.01].
LysoPI Decreased Pro-inflammatory Cytokine Production by LPS-Stimulated BV-2 Microglia
To further explore the effects of LysoPI on microglial inflammatory responses, we next measured cytokine production in LPS-stimulated BV-2 microglia. Cultured BV-2 cells were incubated with LPS with or without LysoPI (20 μM) for 24 h, and the cytokines produced in the medium was determined by Bio-Plex MAGPIX. We measured six pro-inflammatory cytokines and one anti-inflammatory cytokine that are representative of cytokines produced by microglia under inflammatory conditions. The amounts of IL-6 and TNF-α increased by LPS remarkably (Fig. 3c, g). The increased amount of IL-6 by LPS was significantly reduced in the presence of LysoPI by the control level (Fig. 3c); TNF-α was also reduced slightly but not significantly (Fig. 3g). The production of other five cytokines did not change significantly (Fig. 3a, b, d–f).

LysoPI reduced IL-6 production in LPS-stimulated microglia. BV-2 cells were incubated with LPS in the presence or absence of 20 μM LysoPI for 24 h, and then concentrations of cytokines in the medium were measured using bio-Plex MAGPIX. a IL-1β, b IL-2, c IL-6, d IL-10, e IL-12, f INF-γ, g TNF-α. Data are the mean ± S.D. of 4 samples. **p < 0.01, compared to control; #p < 0.05, compared to LPS; Tukey multiple-comparison procedure following one-way ANOVA [c F (3, 12) = 9.45, p < 0.01; g F (3, 12) = 5.47, p < 0.01].
LysoPI Decreased Phagocytic Activity of LPS-Stimulated Microglia
In addition to inflammatory response, phagocytic activity is also important for the microglial cell function. To determine the effects of LysoPI on microglial phagocytic activity, we stimulated BV-2 cells with LPS, with or without LysoPI (20 μM) for 24 h, and the uptake of microbeads was measured. Stimulation with LPS increased the uptake of microbeads by BV-2 cells (Fig. 4a). Furthermore, enhanced phagocytic activity by LPS stimulation was attenuated in the presence of LysoPI by the control level. Also in primary microglia, we examined the effect of LysoPI on phagocytosis to confirm the results in BV-2 cells. The cells were incubated with LPS in the absence or presence of 10 μM LysoPI for 24 h with 0.1% FBS-containing DMEM. Previous reports indicated that serum-free culture for 24 h induces microglial cell death [31] and that serum protein like albumin inhibit the effects of LPLs [28]. Therefore, we supplemented FBS in low concentration which did not cause cell death and inhibit the effects of LysoPI for stimulation of primary microglia. Treatment with LPS increased the uptake of microbeads in primary microglia as well as BV-2 cells (Fig. 4b). Moreover, LPS-induced phagocytic activity was suppressed by LysoPI treatment to the control level, as well as BV-2 cells.

Effect of LysoPI on phagocytic activity of LPS-stimulated microglia. BV-2 cells were incubated with 100 ng/mL LPS together with 20 μM LysoPI with serum-free DMEM for 24 h and then uptake of fluorescent microbeads for 1 h was determined (a). Primary microglia were stimulated with 100 ng/ml LPS in the presence of 10 μM LysoPI with 0.1% FBS-containing DMEM for 24 h, and then phagocytic activity was evaluated using the same method (b). The graph shows the average fluorescent intensity per cell, evaluated using Image J software; data are shown as ratio of control. The photograph shows representative phase contrast (upper) and fluorescent images (lower) at each same site. Data are the mean ± S.D. of 3 (a) or 5 (b) samples. **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to LPS; Tukey multiple-comparison procedure following one-way ANOVA [F (3, 8) = 12.4, p < 0.01]. Scale bar = 20 μm.
The G Protein-Coupled Receptor (GPR) 55-Dependent Pathway Is Involved in LysoPI-Induced Reduction in Phagocytosis by Activated BV-2 Microglia
Recently, several studies have revealed that LPLs elicit various functions, acting on receptors as lipid mediators [8, 9]. It is now widely accepted that LysoPI is a potent endogenous ligand for GPR55, which is known to be highly expressed in primary cultured microglia and BV-2 cells [23]. To assess whether suppression of microglial NO production and phagocytosis by LysoPI is mediated via GPR55, we explored the influence of O-1602, an agonist for GPR55, and CID16020046 (CID), an antagonist for GPR55, on LPS-induced NO production and phagocytic activity in BV-2 cells.
First, to determine whether O-1602 affects microglial NO production as well as LysoPI, BV-2 cells were treated with LPS 100 ng/mL in the presence or absence of O-1602 (30 μM) for 24 h, and then NO production and cell viability were measured. O-1602 remarkably decreased LPS-induced NO production by the control (methyl acetate; MA) level as well as LysoPI (Fig. 5a), without affecting cell viability (Fig. 5b).

Effect of GPR55 agonist, O-1602, on NO production in LPS-stimulated microglia. BV-2 cells were stimulated with LPS together with 30 μM O-1602 for 24 h. Methyl acetate (MA) was used as vehicle, at the concentration less than 0.1%. Nitrite concentration in the medium was determined by fluorescent assay with DAN reagent (a). Cell viability was measured by MTT assay (b). Data are the mean ± S.D. of 4 samples. **p < 0.01, compared to control (MA); ##p < 0.01, compared to LPS (MA); Tukey multiple-comparison procedure following one-way ANOVA [a F (5, 18) = 26.9, p < 0.01; b F (5, 18) = 5.53, p < 0.01].
Second, to investigate the effect of O-1602 on microglial phagocytosis, BV-2 cells were incubated with LPS together with 30 μM O-1602 for 24 h, and the phagocytic activity was evaluated using fluorescent microbeads. O-1602 reduced LPS-induced phagocytosis in BV-2 cells by the control (MA) level (Fig. 6).

Effect of O-1602 on phagocytic activity of LPS-stimulated microglia. BV-2 cells were incubated with LPS in the presence or absence of 30 μM O-1602 for 24 h. After that, fluorescent microbeads were added, then uptake of fluorescent microbeads for 1 h was determined. The results are shown in the photograph and graph as similar to Fig. 4. Data are the mean ± S.D. of 3 samples. **p < 0.01, compared to the control (MA); ##p < 0.01, compared to LPS (MA); Tukey multiple-comparison procedure following one-way ANOVA [F (5, 12) = 23.6, p < 0.01]. Scale bar = 20 μm.
Third, to clarify whether GPR55 is responsible for LysoPI-induced suppression in NO production further using an antagonist, BV-2 cells were pretreated with 100 μM CID 1 h before addition of LPS in the presence or absence of 20 μM LysoPI for 24 h, and then NO production and cell viability were measured. LPS-induced NO production was partly inhibited by the pretreatment with CID (Fig. 7a) without effect on cell viability (Fig. 7b). However, CID did not block the effect of LysoPI on LPS-induced NO production (Fig. 7a).

LysoPI-induced NO suppression was not affected by a GPR55 antagonist, CID. BV-2 cells were treated with 100 μM CID 1 h before LPS stimulation together with 20 μM LysoPI for 24 h. Dimethyl sulfoxide (DMSO) was used as vehicle at the concentration of 0.2%. Nitrite concentration was measured fluorometrically with DAN reagent (a). Cell viability was determined by MTT assay (b). Data are the mean ± S.D. of 3 samples. **p < 0.01, compared to control (DMSO), #p < 0.05, ##p < 0.01, compared to LPS (DMSO); Tukey multiple-comparison procedure following one-way ANOVA [a F (8, 18) = 23.9, p < 0.01; b F (8, 18) = 5.42, p < 0.01].
Finally, to determine whether LysoPI affects microglial phagocytosis via GPR55, BV-2 cells were pretreated with 100 μM CID 1 h before LPS and 20 μM LysoPI stimulation for 24 h. CID completely inhibited the suppressive effect of LysoPI on microglial phagocytic activity by the level in the LysoPI + LPS (DMSO) group (Fig. 8).

Effect of CID on LysoPI-induced reduction in phagocytic activity of LPS-stimulated microglia. BV-2 cells were pretreated with 100 μM CID 1 h before LPS treatment with or without 20 μM LysoPI for 24 h. After that, the uptake of fluorescent microbeads for 1 h was evaluated. The results are shown in the photograph and graph similar to Fig. 4. Data are mean ± S.D. of 3 samples. **p < 0.01, compared to control (DMSO); ##p < 0.01, compared to LPS (DMSO); $$p < 0.01, compared to LysoPI + LPS (DMSO); The Tukey multiple-comparison procedure following one-way ANOVA [F (8, 18) = 31.9, p < 0.01].
DISCUSSION
Several lines of evidence indicate that LPLs mediate inflammation and immunity in the CNS. Among LPLs, LysoPI is an endogenous ligand for GPR55, which is expressed by the microglia and is related to neurodegenerative disorders, such as Parkinson’s and Alzheimer’s disease [7, 20, 21, 23]. Additionally, a significant amount of LysoPI is contained in the rat brain [22]. However, a possible role of LysoPI in neuroinflammation remains unclear. Thus, we explored whether LysoPI can affect the microglial neuroinflammatory response. We demonstrated that LysoPI suppressed the production of NO, generation of ROS, IL-6 release, and phagocytic activity by LPS-stimulated BV-2 microglial cells. Furthermore, we confirmed in primary microglia that LPS-induced intracellular ROS generation and phagocytic activity were also attenuated by LysoPI treatment. In addition, we also investigated the involvement of GPR55 in LysoPI effects and revealed that a GPR55 agonist reduced the LPS-stimulated phagocytic activity, and that an antagonist for GPR55 inhibited the effect of LysoPI on phagocytic activity in LPS-stimulated BV-2 cells. These results suggest that LysoPI reduces the microglial inflammatory responses and the suppressive effect of LysoPI on microglial phagocytosis is mediated by GPR55.
The microglia are activated by injury or inflammation in the CNS and play an important role in maintaining CNS homeostasis by phagocytosis of debris and production of cytokines [1,2,3,4]. However, the microglia damage and phagocytose neurons by producing excess pro-inflammatory substances such as NO, ROS, TNF-α, and IL-1β, when they were hyper-activated [1, 2, 5]. This study demonstrated that LysoPI suppressed NO and ROS production, inflammatory cytokine release, and phagocytosis by LPS-stimulated BV-2 microglia. It has been reported that LysoPI exerts a neuroprotective effect in the microglia against neuronal excitotoxicity by N-methyl-D-aspartate, a selective agonist for glutamate receptor [24]. Therefore, our findings suggest that LysoPI may protect neurons from excitotoxicity by reducing the production of pro-inflammatory factors and excess phagocytic activity in the microglia.
ROS function as signal molecules in neuroinflammation, while excessive generation of ROS causes oxidative stress in neurons and glia resulting in neuronal damage or degeneration [32, 33]. We found that LysoPI attenuated LPS-induced intracellular ROS generation both in BV-2 (Fig. 2a) and primary microglia (Fig. 2b), suggesting that LysoPI suppressed oxidative stress by ROS. Moreover, production of IL-6 was also reduced by LysoPI (Fig. 3c). Intracellular ROS are thought to be generated by mitochondrial electron transport chain or NADPH oxidase (NOX) or other pathways [33]. Several lines of evidence have indicated that NOX-dependent ROS stimulates the production of inflammatory cytokine, including IL-1β, TNF-α, and IL-6 [34, 35]. Therefore, the suppression of IL-6 production by LysoPI may be due to the decrease in ROS generation. Balenga et al. [36] indicated that LysoPI reduced ROS generation in human neutrophils activated with 2-arachidonoylglycerol, a CB2 receptor agonist through the GPR55 pathway. Thus, LysoPI may have attenuated LPS-induced ROS generation in this study via GPR55. On the other hand, it has also been reported that LysoPI induced ROS generation in human endothelial cells [37] and enhanced ROS generation in activated human neutrophils [38]. This contradiction between our results and previous findings may occur due to the differences in LysoPI acyl chain length and degree of unsaturation. Several lines of evidence have indicated that LPLs trigger different signaling pathways, which results in varied functional outcomes according to their acyl chain length and degree of unsaturation [39,40,41,42]. In fact, the acyl chain length of LysoPI used in the present study was different from that used in the previous report [37] although the other study [38] did not show details about the structure of LysoPI.
Recently, a growing body of evidence has revealed that LysoPI is a ligand for GPR55 [7]. In addition, it has been reported that GPR55 is highly expressed in primary cultured microglial cells and BV-2 cells [23]. It has also been suggested that LysoPI acts on GPR119 and potassium channels [7, 8]. Being consistent with the results of LysoPI (Fig. 1a), O-1602, an agonist for GPR55, suppressed LPS-induced NO production (Fig. 5a). LPS-induced microglial phagocytic activity was also reduced by co-stimulation with O-1602 (Fig. 6) as well as LysoPI (Fig. 4a). In addition, pretreatment with CID, an antagonist for GPR55, reversed the suppressive effect of LysoPI on LPS-induced microglial phagocytosis (Fig. 8), whereas LysoPI-induced decrease in NO production was not affected by CID (Fig. 7a). These results suggest that LysoPI modulates microglial phagocytic activity through the GPR55-dependent pathway. On the other hand, it is also suggested that LysoPI may suppress LPS-induced NO production through a pathway other than the GPR55-dependent pathway. For example, LysoPI can bind directly to membrane proteins, such as ion channels, or create a plasma membrane curvature by its insertion into the outer leaflet of the lipid bilayer, which may result in changes in the properties of the cell membrane and proteins including ion channels [7, 8, 43]. In particular, it has been indicated that LysoPI affects the activity of various types of ion channels, particularly potassium channels, by a receptor-independent manner in non-microglial cells such as endothelial cells [8, 44,45,46,47]. In addition, accumulating evidence indicated that activation of the potassium channel by LPS stimulation results in enhanced production of NO in the microglia [48, 49]. Therefore, LysoPI-induced reduction in NO production in this study might be due to the inactivation of potassium channels. As for the effects of CID, someone may have questions about why pre-stimulation with CID, an antagonist for GPR55, significantly suppressed LPS-induced NO production by BV-2 microglial cells even without LysoPI in this study (Fig. 7a). The same finding was also reported in the rat primary microglia by Malek et al. [50]. In addition, Saliba et al. [51] indicated that GPR55 antagonism resulted in anti-inflammation in the primary microglia. Alhouayek et al. [7] reported that nuclear factor-kappa B (NF-κB) is activated through ERK in the signal pathway below GPR55. NF-κB activation leads to the enhancement of NO production via increase in iNOS expression [2, 52]. Our results and these previous findings suggest that the activation of the GPR55 pathway may induce neuroinflammation in the activated microglia. However, it is necessary to consider the pharmacological property of CID, which has the potential to antagonize other receptors like GPR18 expressed in the microglia [50, 53, 54]. Therefore, further studies are needed to clarify the function of GPR55 in neuroinflammation, particularly in the microglia. Taken together, our findings suggest that LysoPI decreased microglial phagocytosis via the GPR55-dependent pathway, while the suppressive effect of LysoPI on NO production may depend on receptor pathways other than GPR55 or interaction with the cell membrane or protein.
LysoPI suppressed LPS-induced phagocytosis in BV-2 cells (Fig. 4a) and primary microglia (Fig. 4b). In addition, a synthetic agonist for GPR55, O-1602, also reduced LPS-induced microglial phagocytosis (Fig. 6) and CID, an antagonist for GPR55, completely reversed the suppressive effect of LysoPI on LPS-induced phagocytosis (Fig. 8). These results suggest that LysoPI reduces microglial phagocytosis via GPR55. Phagocytosis by microglia is provoked by several factors including LPS, apoptotic debris, and extracellular nucleotides through respective receptors expressed on the cell surface [55]. Above all, LPS induces microglial phagocytosis by activation of the Toll-like receptor 4 signaling pathway including p38 mitogen-activated protein kinase (p38 MAPK) and Cdc42/Rac signaling [55]. Phagocytosis by the microglia is also provoked by activation of ERK via triggering receptor expressed on myeloid cells 2 receptor [55]. The GPR55 signaling pathway induces activation of p38 MAPK through Rac1/Cdc42 [7]. Moreover, ERK can be activated by stimulation of GPR55 [7]. These downstream signals of GPR55 may accelerate further the microglial phagocytosis. Furthermore, in macrophages, GPR55 activation by palmitoylethanolamide enhanced the expression of phagocytosis-related receptors and efferocytosis [56]. However, these previous findings [7, 56] contradict our results. Pietr et al. [23] demonstrated that the expression level of GPR55 changes according to the stimulation agent and cell species. For example, INF-γ upregulated the expression of GPR55, but LPS downregulated it. Thus, a possible explanation for this contradiction might be that the cell species and agent for cellular activation used in the study were different between the previous study and our study.
Although the mechanism for suppression in phagocytosis by LysoPI through GPR55 is unclear, there are some possibilities to explain this issue. One possible explanation for this is that GPR55 may interact with other proteins including receptors. Recently, a growing body of evidence has suggested that heteromerization of GPR55 and cannabinoid 2 (CB2) receptor modulates receptor signaling with each other [7, 57, 58]. Both of these receptors are expressed by the primary microglia and BV-2 cells [23, 59, 60]. For instance, Balenga et al. [36] demonstrated that the GPR55 activation by LysoPI could lead to both the acceleration and the inhibition of the CB2 receptor-mediated cellular response in neutrophils. As for functions of the CB2 receptor, multiple lines of evidence suggest that the CB2 receptor induces anti-inflammatory responses in the microglia, for example, reduction in the release of inflammatory factors, pro-inflammatory cytokines, and ROS [59, 60]. It was also demonstrated that the synthetic agonist of the CB2 receptor attenuated basal phagocytic activity by BV-2 microglia [61]. Therefore, LysoPI might reduce microglial phagocytosis by activating the CB2 receptor through activation of GPR55. In addition, it has also been reported that GPR55 antagonism induces cross-antagonism of the CB2 receptor [58]. Thus, inhibition of LysoPI-induced reduction in microglial phagocytosis by CID, an antagonist for GPR55, may be due to the cross-antagonism of the CB2 receptor by CID. Additionally, Ailte et al. [43] demonstrated that LysoPI inhibited macrophage endocytosis by reducing clathrin-coated pits and caveolae through changes in the properties of cell membranes. Therefore, we cannot rule out the possibility that LysoPI suppressed microglial phagocytosis through direct alteration of the property of the plasma membrane by insertion of LysoPI into the lipid bilayer. Further study should be undertaken to elucidate the mechanism by which LysoPI suppressed LPS-induced microglial phagocytosis via GPR55.
Our results revealed that LysoPI had anti-inflammatory effects on activated microglia and the effect on phagocytosis was exerted through the GPR55 pathway. Recent studies have found that GPR55 can be involved in several brain functions like social interaction, memory, and anxiety [7, 62]. It has also been revealed that GPR55 can be a therapeutic target for the treatment of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [7, 20, 21]. For instance, a synthetic cannabidiol isomer is a GPR55 agonist, which relieved motor dysfunctions in Parkinson disease model mice induced with haloperidol [20]. Thus, our results suggest that microglial GPR55 may be partly responsible for the therapeutic effect of GPR55. Further study is required to demonstrate whether the therapeutic effect of GPR55-targeting treatment for neurodegenerative disorders depends on microglial GPR55.
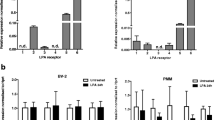
In this study, we assessed the effect of LysoPI on intracellular ROS generation and phagocytic activity in primary microglia as well as BV-2 cells. On the other hand, in primary microglia, we were not able to observe LPS-induced NO production and its suppression by LysoPI because of the low cell viability and insufficient potentiation of NO production. It was reported that serum-free culture for 24 h can induce microglial cell death [31]. It was also found that LPS induced NO production in a serum-dependent manner in mixed glial cells [63]. Thus, we were not able to observe the LPS-induced NO production and its suppression by LysoPI in primary microglia at 24 h under serum-deprived condition. Furthermore, we did not measure cytokine production in primary microglia because we considered that the effects of LysoPI on inflammatory responses in BV-2 cells and primary microglia were comparable according to the results of ROS generation and phagocytosis. We used 10 μM LysoPI in primary microglia that are lower than in BV-2 cells (20 μM). We considered that it is preferable to use LysoPI at lower concentration by which inflammatory responses in primary microglia can be affected because LysoPI at low concentration can have fewer influences in cell viability than at high concentration. Therefore, we used 10 μM LysoPI in primary microglia. The reason why 10 μM LysoPI was effective in primary microglia may be explained by differences in receptor expression. It has been reported that several candidate receptors for LysoPI are expressed both in primary microglia and BV-2 cells and that their expression levels are different between both cells [7, 8, 23, 64]. It was also found that BV-2 cells and primary microglia expressed different types of lysophosphatidic acid (LPA) receptor, respectively, and LPA-induced ROS generation in primary microglia was observed at a shorter stimulation time than that in BV-2 cells [65]. Thus, the sensitivity to LysoPI may be higher in primary microglia than BV-2 cells because of the differences in the expression levels or types of receptor expressed in them.
We did not detect whether CID inhibits the effect of O-1602 on LPS-induced microglial inflammatory response in this study. In previous studies, O-1602 as GPR55 agonist and CID as GPR55 antagonist were commonly used, respectively [66,67,68,69]. In addition, it was found that O-1602 suppressed IL-1β-induced cytokine receptor expression in neural stem cells and the suppressive effect of O-1602 was inhibited by CID [67]. Thus, it is supposed that the suppressive effect of O-1602 on LPS-induced NO production can be inhibited by CID treatment. Although we did not reveal the effect of O-1602 on microglial cytokine release, previous studies supposed that O-1602 can reduce pro-inflammatory cytokine, such as IL-6, release. It has been demonstrated that neurotoxicity by activated microglia, which was mediated by inflammatory factors, including cytokine released from microglia, was suppressed by O-1602 [70]. Furthermore, a previous report demonstrated that O-1602 treatment decreased the level of pro-inflammatory cytokines including IL-6 in tissue or plasma of pancreatitis and colitis [71, 72]. Additionally, we demonstrated that O-1602 has anti-inflammatory effects in LPS-stimulated microglia. Therefore, we assume that O-1602 can reduce pro-inflammatory cytokine production in microglia.
In conclusion, we demonstrated that LysoPI suppressed neuroinflammation mediated by the microglia, resulting in the reduction of NO, iNOS, ROS, IL-6, and phagocytic activity. The effects of LysoPI on microglia are, at least partly, dependent on the GPR55 pathway. These findings suggest that the previously reported [24] neuroprotective effects of LysoPI are exerted by attenuating the microglia-mediated inflammation. In addition, our findings suggest that the therapeutic effect of GPR55-targeting treatment for neurodegenerative diseases may partly depend on microglial GPR55.
Abbreviations
- CB2 :
-
Cannabinoid 2
- DAN:
-
2,3-Diaminonaphthalene
- DCF:
-
2′,7′-Dichlorofluorescein
- DMEM:
-
Dulbecco’s modified Eagle medium
- DMSO:
-
Dimethyl sulfoxide
- ERK:
-
Extracellular signal-regulated kinase
- FBS:
-
Fetal bovine serum
- GPR:
-
G protein-coupled receptor
- H2DCFDA:
-
2′,7′-Dichlorodihydrofluorescein diacetate
- IL:
-
Interleukin
- INF-γ:
-
Interferon-γ
- iNOS:
-
Inducible nitric oxide synthase
- LPLs:
-
Lysophospholipids
- LPS:
-
Lipopolysaccharide
- LysoPI:
-
Lysophosphatidylinositol
- MA:
-
Methyl acetate
- MTT:
-
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide
- NF-κB:
-
Nuclear factor-kappa B
- NO:
-
Nitric oxide
- ROS:
-
Reactive oxygen species
- TNF-α:
-
Tumor necrosis factor-α
References
Kreutzberg, G.W. 1996. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences 19: 312–318. https://doi.org/10.1016/0166-2236(96)10049-7.
Nakamura, Y. 2002. Regulating factors for microglial activation. Biological and Pharmaceutical Bulletin 25: 945–953. https://doi.org/10.1248/bpb.25.945.
Block, M.L., L. Zecca, and J. Hong. 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience 8: 57–69. https://doi.org/10.1038/nrn2038.
Neniskyte, U., J.J. Neher, and G.C. Brown. 2011. Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. Journal of Biological Chemistry 286: 39904–39913. https://doi.org/10.1074/jbc.M111.267583.
Brown, G.C., and A. Vilalta. 2015. How microglia kill neurons. Brain Research 1628: 288–297. https://doi.org/10.1016/j.brainres.2015.08.031.
Aoki, J., Y. Nagai, H. Hosono, K. Inoue, and H. Arai. 2002. Structure and function of phosphatidylserine-specific phospholipase A1. Biochimica et Biophysica Acta 1582 (1–3): 26–32. https://doi.org/10.1016/s1388-1981(02)00134-8.
Alhouayek, M., J. Masquelier, and G.G. Muccioli. 2018. Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends in Pharmacological Sciences 39 (6): 586–604. https://doi.org/10.1016/j.tips.2018.02.011.
Grzelczyk, A., and E. Gendaszewska-Darmach. 2013. Novel bioactive glycerol-based lyosphospholipids: new data ― new insight into their function. Biochimie 95: 667–679. https://doi.org/10.1016/j.biochi.2012.10.009.
Kihara, Y., H. Mizuno, and J. Chun. 2015. Lysophospholipid receptors in drug discovery. Experimental Cell Research 333 (2): 171–177. https://doi.org/10.1016/j.yexcr.2014.11.020.
Zhao, C., M.J. Fernandes, G.D. Prestwich, M. Turgeon, J. Di Battista, T. Clair, P.E. Poubelle, and S.G. Bourgoin. 2008. Regulation of lysophosphatidic acid receptor expression and function in human synoviocytes: implications for rheumatoid arthritis? Molecular Pharmacology 73 (2): 587–600. https://doi.org/10.1124/mol.107.038216.
Bektas, M., M.L. Allende, B.G. Lee, W. Chen, M.J. Amar, A.T. Remaley, J.D. Saba, and R.L. Proia. 2010. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. Journal of Biological Chemistry 285 (14): 10880–10889. https://doi.org/10.1074/jbc.M109.081489.
Vladykovskaya, E., E. Ozhegov, J.D. Hoetker, Z. Xie, Y. Ahmed, J. Suttles, S. Srivastava, A. Bhatnagar, and O.A. Barski. 2011. Reductive metabolism increases the proinflammatory activity of aldehyde phospholipids. Journal of Lipid Research 52 (12): 2209–2225. https://doi.org/10.1194/jlr.M013854.
Hung, N.D., M.R. Kim, and D.E. Sok. 2011. 2-Polyunsaturated acyl lysophosphatidylethanolamine attenuates inflammatory response in zymosan A-induced peritonitis in mice. Lipids 46: 893–906. https://doi.org/10.1007/s11745-011-3589-2.
Nishikawa, M., M. Kurano, H. Ikeda, J. Aoki, and Y. Yatomi. 2015. Lysophosphatidylserine has bilateral effects on macrophages in the pathogenesis of atherosclerosis. Journal of Atherosclerosis and Thrombosis 22 (5): 518–526. https://doi.org/10.5551/jat.25650.
Henstridge, C.M., N.A. Balenga, L.A. Ford, R.A. Ross, M. Waldhoer, and A.J. Irving. 2009. The GPR55 ligand L-α-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB Journal 23 (1): 183–193. https://doi.org/10.1096/fj.08-108670.
Anavi-Goffer, S., G. Baillie, A.J. Irving, J. Gertsch, I.R. Greig, R.G. Pertwee, and R.A. Ross. 2011. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. Journal of Biological Chemistry 287 (1): 91–104. https://doi.org/10.1074/jbc.M111.296020.
Frasch, S.C., and D.L. Bratton. 2012. Emerging roles for lysophosphatidylserine in resolution of inflammation. Progress in Lipid Research 51 (3): 199–207. https://doi.org/10.1016/j.plipres.2012.03.001.
Kamat, S.S., K. Camara, W.H. Parsons, D.H. Chen, M.M. Dix, T.D. Bird, A.R. Howell, and B.F. Cravatt. 2015. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nature Chemical Biology 11 (2): 164–171. https://doi.org/10.1038/nchembio.1721.
Wang, X., Y.F. Li, G. Nanayakkara, Y. Shao, B. Liang, L. Cole, W.Y. Yang, X. Li, R. Cueto, J. Yu, H. Wang, and X.F. Yang. 2016. Lysophospholipid receptors, as novel conditional danger receptors and homeostatic receptors modulate inflammation-novel paradigm and therapeutic potential. Journal of Cardiovascular Translational Research 9 (4): 343–359. https://doi.org/10.1007/s12265-016-9700-6.
Celorrio, M., E. Rojo-Bustamante, D. Fernández-Suárez, E. Sáez, A. Estella-Hermoso de Mendoza, C.E. Müller, M.J. Ramírez, J. Oyarzábal, R. Franco, and M.S. Aymerich. 2017. GPR55: a therapeutic target for Parkinson’s disease? Neuropharmacology 125: 319–332. https://doi.org/10.1016/j.neuropharm.2017.08.017.
Haque, M.E., I.S. Kim, M. Jakaria, M. Akther, and D.K. Choi. 2018. Importance of GPCR-mediated microglial activation in Alzheimer’s disease. Frontiers in Cellular Neuroscience 12: 258. https://doi.org/10.3389/fncel.2018.00258.
Oka, S., T. Toshida, K. Maruyama, K. Nakajima, A. Yamashita, and T. Sugiura. 2009. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: a possible natural ligand for GPR55. Journal of Biochemistry 145 (1): 13–20. https://doi.org/10.1093/jb/mvn136.
Pietr, M., E. Kozela, R. Levy, N. Rimmerman, Y.H. Lin, N. Stella, Z. Vogel, and A. Juknat. 2009. Differential changes in GPR55 during microglial cell activation. FEBS Letters 583 (12): 2071–2076. https://doi.org/10.1016/j.febslet.2009.05.028.
Kallendrusch, S., S. Kremzow, M. Nowicki, U. Grabiec, R. Winkelmann, A. Benz, and M. Koch. 2013. The G protein-coupled receptor 55 ligand L-α-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia 61 (11): 1822–1831. https://doi.org/10.1002/glia.22560.
Si, Q., Y. Nakamura, and K. Kataoka. 2000. A serum factor enhances production of nitric oxide and tumor necrosis factor-α from cultured microglia. Experimental Neurology 162 (1): 89–97. https://doi.org/10.1006/exnr.2000.7334.
Horvath, R.J., N. Nutile-McMenemy, M.S. Alkaitis, and J.A. Deleo. 2008. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. Journal of Neurochemistry 107 (2): 557–569. https://doi.org/10.1111/j.1471-4159.2008.05633.x.
de Jong, E.K., A.H. de Haas, N. Brouwer, H.R. van Weering, M. Hensens, I. Bechmann, P. Pratley, E. Wesseling, H.W. Boddeke, and K. Biber. 2008. Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. Journal of Neurochemistry 105 (5): 1726–1736. https://doi.org/10.1111/j.1471-4159.2008.05267.x.
Kim, Y.L., Y.J. Im, N.C. Ha, and D.S. Im. 2006. Albumin inhibits cytotoxic activity of lysophosphatidylcholine by direct binding. Prostaglandins & Other Lipid Mediators 83 (1-2): 130–138. https://doi.org/10.1016/j.prostaglandins.2006.10.006.
Takano, K., K. Sugita, M. Moriyama, K. Hashida, S. Hibino, T. Choshi, R. Murakami, M. Yamada, H. Suzuki, O. Hori, and Y. Nakamura. 2011. A dibenzoylmethane derivative protects against hydrogen peroxide-induced cell death and inhibits lipopolysaccharide-induced nitric oxide production in cultured rat astrocytes. Journal of Neuroscience Research 89: 955–965. https://doi.org/10.1002/jnr.22617.
Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72 (1–2): 248–254. https://doi.org/10.1006/abio.1976.9999.
Koyama, Y., Y. Kimura, Y. Yoshioka, D. Wakamatsu, R. Kozaki, H. Hashimoto, T. Matsuda, and A. Baba. 2000. Serum-deprivation induces cell death of rat cultured microglia accompanied with expression of Bax protein. Japanese Journal of Pharmacology 83 (4): 351–354. https://doi.org/10.1254/jjp.83.351.
Hsieh, H.L., and C.M. Yang. 2013. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Research International 2013: 484613–484618. https://doi.org/10.1155/2013/484613.
Mittal, M., M.R. Siddiqui, K. Tran, S.P. Reddy, and A.B. Malik. 2014. Reactive oxygen species in inflammation and tissue injury. Antioxidants & Redox Signaling 20 (7): 1126–1167. https://doi.org/10.1089/ars.2012.5149.
Qin, L., Y. Liu, T. Wang, S.J. Wei, M.L. Block, B. Wilson, B. Liu, and J.S. Hong. 2004. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. Journal of Biological Chemistry 279 (2): 1415–1421. https://doi.org/10.1074/jbc.M307657200.
Li, B., K. Bedard, S. Sorce, B. Hinz, M. Dubois-Dauphin, and K.H. Krause. 2009. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. Journal of Innate Immunity 1 (6): 570–581. https://doi.org/10.1159/000235563.
Balenga, N.A., E. Aflaki, J. Kargl, W. Platzer, R. Schröder, S. Blättermann, E. Kostenis, A.J. Brown, A. Heinemann, and M. Waldhoer. 2011. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Research 21 (10): 1452–1469. https://doi.org/10.1038/cr.2011.60.
Li, X., L. Wang, P. Fang, Y. Sun, X. Jiang, H. Wang, and X.F. Yang. 2018. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. Journal of Biological Chemistry 293 (28): 11033–11045. https://doi.org/10.1074/jbc.RA118.002752.
Ginsburg, I., P.A. Ward, and J. Varani. 1989. Lysophosphatides enhance superoxide responses of stimulated human neutrophils. Inflammation 13 (2): 163–174. https://doi.org/10.1007/bf00924787.
Ojala, P.J., T.E. Hirvonen, M. Hermansson, P. Somerharju, and J. Parkkinen. 2007. Acyl chain-dependent effect of lysophosphatidylcholine on human neutrophils. Journal of Leukocyte Biology 82 (6): 1501–1509. https://doi.org/10.1189/jlb.0507292.
Brkić, L., M. Riederer, W.F. Graier, R. Malli, and S. Frank. 2012. Acyl chain-dependent effect of lysophosphatidylcholine on cyclooxygenase (COX)-2 expression in endothelial cells. Atherosclerosis 224 (2): 348–354. https://doi.org/10.1016/j.atherosclerosis.2012.07.038.
Rao, S.P., M. Riederer, M. Lechleitner, M. Hermansson, G. Desoye, S. Hallström, W.F. Graier, and S. Frank. 2013. Acyl chain-dependent effect of lysophosphatidylcholine on endothelium-dependent vasorelaxation. PLoS One 8 (5): e65155. https://doi.org/10.1371/journal.pone.0065155.
Tokizane, K., H. Konishi, K. Makide, H. Kawana, S. Nakamuta, K. Kaibuchi, T. Ohwada, J. Aoki, and H. Kiyama. 2017. Phospholipid localization implies microglial morphology and function via Cdc42 in vitro. Glia 65 (5): 740–755. https://doi.org/10.1002/glia.23123.
Ailte, I., A.B. Lingelem, A.S. Kvalvaag, S. Kavaliauskiene, A. Brech, G. Koster, P.G. Dommersnes, J. Bergan, T. Skotland, and K. Sandvig. 2017. Exogenous lysophospholipids with large head groups perturb clathrin-mediated endocytosis. Traffic 18 (3): 176–191. https://doi.org/10.1111/tra.12468.
Amorós, I., A. Barana, R. Caballero, R. Gómez, L. Osuna, M.P. Lillo, J. Tamargo, and E. Delpón. 2010. Endocannabinoids and cannabinoid analogues block human cardiac Kv4.3 channels in a receptor-independent manner. Journal of Molecular and Cellular Cardiology 48 (1): 201–210. https://doi.org/10.1016/j.yjmcc.2009.07.011.
Bondarenko, A., M. Waldeck-Weiermair, S. Naghdi, M. Poteser, R. Malli, and W.F. Graier. 2010. GPR55-dependent and -independent ion signalling in response to lysophosphatidylinositol in endothelial cells. British Journal of Pharmacology 161 (2): 308–320. https://doi.org/10.1111/j.1476-5381.2010.00744.x.
Bondarenko, A.I., R. Malli, and W.F. Graier. 2011. The GPR55 agonist lysophosphatidylinositol directly activates intermediate-conductance Ca2+-activated K+ channels. Pflügers Archiv: European Journal of Physiology 462 (2): 245–255. https://doi.org/10.1007/s00424-011-0977-7.
Karpińska, O., M. Baranowska-Kuczko, B. Malinowska, M. Kloza, M. Kusaczuk, A. Gęgotek, P. Golec, I. Kasacka, and H. Kozłowska. 2018. Mechanisms of L-alpha-lysophosphatidylinositol-induced relaxation in human pulmonary arteries. Life Sciences 192: 38–45. https://doi.org/10.1016/j.lfs.2017.11.020.
Kaushal, V., P.D. Koeberle, Y. Wang, and L.C. Schlichter. 2007. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. The Journal of Neuroscience 27 (1): 234–244. https://doi.org/10.1523/JNEUROSCI.3593-06.2007.
Nguyen, H.M., E.M. Grössinger, M. Horiuchi, K.W. Davis, L.W. Jin, I. Maezawa, and H. Wulff. 2017. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65 (1): 106–121. https://doi.org/10.1002/glia.23078.
Malek, N., K. Popiolek-Barczyk, J. Mika, B. Przewlocka, and K. Starowicz. 2015. Anandamide, acting via CB2 receptors, alleviates LPS-induced neuroinflammation in rat primary microglial cultures. Neural Plasticity 2015: 130639–130610. https://doi.org/10.1155/2015/130639.
Saliba, S.W., H. Jauch, B. Gargouri, A. Keil, T. Hurrle, N. Volz, F. Mohr, M. van der Stelt, S. Bräse, and B.L. Fiebich. 2018. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. Journal of Neuroinflammation 15 (1): 322. https://doi.org/10.1186/s12974-018-1362-7.
Lieb, K., S. Engels, and B.L. Fiebich. 2003. Inhibition of LPS-induced iNOS and NO synthesis in primary rat microglial cells. Neurochemistry International 42: 131–137. https://doi.org/10.1016/s0197-0186(02)00076-1.
McHugh, D. 2012. GPR18 in microglia: Implications for the CNS and endocannabinoid system signaling. British Journal of Pharmacology 167 (8): 1575–1582. https://doi.org/10.1111/j.1476-5381.2012.02019.x.
Reyes-Resina, I., G. Navarro, D. Aguinaga, E.I. Canela, C.T. Schoeder, M. Załuski, K. Kieć-Kononowicz, C.A. Saura, C.E. Müller, and R. Franco. 2018. Molecular and functional interaction between GPR18 and cannabinoid CB2 G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochemical Pharmacology 157: 169–179. https://doi.org/10.1016/j.bcp.2018.06.001.
Fu, R., Q. Shen, P. Xu, J.J. Luo, and Y. Tang. 2014. Phagocytosis of microglia in the central nervous system diseases. Molecular Neurobiology 49 (3): 1422–1434. https://doi.org/10.1007/s12035-013-8620-6.
Rinne, P., R. Guillamat-Prats, M. Rami, L. Bindila, L. Ring, L.P. Lyytikäinen, E. Raitoharju, N. Oksala, T. Lehtimäki, C. Weber, E.P.C. van der Vorst, and S. Steffens. 2018. Palmitoylethanolamide promotes a proresolving macrophage phenotype and attenuates atherosclerotic plaque formation. Arteriosclerosis, Thrombosis, and Vascular Biology 38 (11): 2562–2575. https://doi.org/10.1161/ATVBAHA.118.311185.
Balenga, N.A., E. Martínez-Pinilla, J. Kargl, R. Schröder, M. Peinhaupt, W. Platzer, Z. Bálint, M. Zamarbide, I.G. Dopeso-Reyes, A. Ricobaraza, J.M. Pérez-Ortiz, E. Kostenis, M. Waldhoer, A. Heinemann, and R. Franco. 2014. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. British Journal of Pharmacology 171 (23): 5387–5406. https://doi.org/10.1111/bph.12850.
Moreno, E., C. Andradas, M. Medrano, M.M. Caffarel, E. Pérez-Gómez, S. Blasco-Benito, M. Gómez-Cañas, M.R. Pazos, A.J. Irving, C. Lluís, E.I. Canela, J. Fernández-Ruiz, M. Guzmán, P.J. McCormick, and C. Sánchez. 2014. Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. Journal of Biological Chemistry 289 (32): 21960–21972. https://doi.org/10.1074/jbc.M114.561761.
Stella, N. 2009. Endocannabinoid signaling in microglial cells. Neuropharmacology 56 Suppl 1: 244–253. https://doi.org/10.1016/j.neuropharm.2008.07.037.
Mecha, M., F.J. Carrillo-Salinas, A. Feliú, L. Mestre, and C. Guaza. 2016. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacology and Therapeutics 166: 40–55. https://doi.org/10.1016/j.pharmthera.2016.06.011.
Hassan, S., K. Eldeeb, P.J. Millns, A.J. Bennett, S.P. Alexander, and D.A. Kendall. 2014. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. British Journal of Pharmacology 171 (9): 2426–2439. https://doi.org/10.1111/bph.12615.
Marichal-Cancino, B.A., A. Fajardo-Valdez, A.E. Ruiz-Contreras, M. Mendez-Díaz, and O. Prospero-García. 2017. Advances in the physiology of GPR55 in the central nervous system. Current Neuropharmacology 15 (5): 771–778. https://doi.org/10.2174/1570159X14666160729155441.
Cooper, C.L., G.H. Jeohn, P. Tobias, and J.S. Hong. 2002. Serum-dependence of LPS-induced neurotoxicity in rat cortical neurons. Annals of the New York Academy of Sciences 962: 306–317. https://doi.org/10.1111/j.1749-6632.2002.tb04076.x.
McHugh, D., S.S. Hu, N. Rimmerman, A. Juknat, Z. Vogel, J.M. Walker, and H.B. Bradshaw. 2010. N-Arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neuroscience 11: 44. https://doi.org/10.1186/1471-2202-11-44.
Plastira, I., E. Bernhart, M. Goeritzer, H. Reicher, V.B. Kumble, N. Kogelnik, A. Wintersperger, A. Hammer, S. Schlager, K. Jandl, A. Heinemann, D. Kratky, E. Malle, and W. Sattler. 2016. 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. Journal of Neuroinflammation 13 (1): 205. https://doi.org/10.1186/s12974-016-0701-9.
Hill, J.D., V. Zuluaga-Ramirez, S. Gajghate, M. Winfield, and Y. Persidsky. 2018. Activation of GPR55 increases neural stem cell proliferation and promotes early adult hippocampal neurogenesis. British Journal of Pharmacology 175 (16): 3407–3421. https://doi.org/10.1111/bph.14387.
Hill, J.D., V. Zuluaga-Ramirez, S. Gajghate, M. Winfield, U. Sriram, S. Rom, and Y. Persidsky. 2019. Activation of GPR55 induces neuroprotection of hippocampal neurogenesis and immune responses of neural stem cells following chronic, systemic inflammation. Brain, Behavior, and Immunity 76: 165–181. https://doi.org/10.1016/j.bbi.2018.11.017.
Stančić, A., K. Jandl, C. Hasenöhrl, F. Reichmann, G. Marsche, R. Schuligoi, A. Heinemann, M. Storr, and R. Schicho. 2015. The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterology and Motility 27 (10): 1432–1445. https://doi.org/10.1111/nmo.12639.
Brown, A.J., I. Castellano-Pellicena, C.P. Haslam, P.L. Nichols, and S.J. Dowell. 2018. Structure-activity relationship of the GPR55 antagonist, CID16020046. Pharmacology 102 (5–6): 324–331. https://doi.org/10.1159/000493490.
Janefjord, E., J.L. Mååg, B.S. Harvey, and S.D. Smid. 2014. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cellular and Molecular Neurobiology 34 (1): 31–42. https://doi.org/10.1007/s10571-013-9984-x.
Li, K., J.Y. Feng, Y.Y. Li, B. Yuece, X.H. Lin, L.Y. Yu, Y.N. Li, Y.J. Feng, and M. Storr. 2013. Anti-inflammatory role of cannabidiol and O-1602 in cerulein-induced acute pancreatitis in mice. Pancreas 42 (1): 123–129. https://doi.org/10.1097/MPA.0b013e318259f6f0.
Wei, D., H. Wang, J. Yang, Z. Dai, R. Yang, S. Meng, Y. Li, and X. Lin. 2020. Effects of O-1602 and CBD on TNBS-induced colonic disturbances. Neurogastroenterology and Motility 32 (3): e13756. https://doi.org/10.1111/nmo.13756.
Funding
This work was funded in part by JSPS KAKENHI Grant No. JP18K05999 to Y.N., JP17K15390 to K.T., and JP17K08127 to M.M.
Author information
Authors and Affiliations
Contributions
T.M. and M.M. contributed to the study concept and design, analysis, and drafting of the manuscript. K.T. participated in the data analysis and drafting of the manuscript. Y.N. provided helpful discussion, data interpretation, and manuscript review. All of the authors edited the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Minamihata, T., Takano, K., Moriyama, M. et al. Lysophosphatidylinositol, an Endogenous Ligand for G Protein-Coupled Receptor 55, Has Anti-inflammatory Effects in Cultured Microglia. Inflammation 43, 1971–1987 (2020). https://doi.org/10.1007/s10753-020-01271-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-020-01271-4