
Abstract
Microglia plays a prominent role in the brain’s inflammatory response to injury or infection by migrating to affected locations and secreting inflammatory molecules. However, hyperactivated microglial is neurotoxic and plays critical roles in the pathogenesis of neurodegenerative diseases. Pristimerin, a naturally occurring triterpenoid, possesses antitumor, antioxidant, and anti-inflammatory activities. However, the effect and the molecular mechanism of pristimerin against lipopolysaccharide (LPS)-induced neurotoxicity in microglia remain to be revealed. In the present study, using BV-2 microglial cultures, we investigated whether pristimerin modifies neurotoxicity after LPS stimulation and which intracellular pathways are involved in the effect of pristimerin. Here we show that pristimerin markedly suppressed the release of Regulated on Activation, Normal T Expressed and Secreted (RANTES), transforming growth factor-β1 (TGF-β1), IL-6, tumor necrosis factor-α (TNF-α), and nitric oxide (NO). Pristimerin also significantly inhibited migration of BV-2 microglia and alleviated the death of neuron-like PC12 cell induced by the conditioned medium from LPS-activated BV-2 microglial cells. Moreover, pristimerin reduced the expression and interaction of TNF Receptor-Associated Factor 6 (TRAF6) and Interleukin-1 Receptor-Associated Kinases (IRAK1), limiting TGF-beta activating kinase 1 (TAK1) activation, and resulting in an inhibition of IKKα/β/NF-κB and MKK7/JNK/AP-1 signaling pathway in LPS-activated BV-2 microglia. Taken together, the anti-neurotoxicity action of pristimerin is mediated through the inhibition of TRAF6/IRAK1/TAK1 interaction as well as the related pathways: IKKα/β/NF-κB and MKK7/JNK/AP-1 signaling pathways. These findings may suggest that pristimerin might serve as a new therapeutic agent for treating hyperactivated microglial induced neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microglia are immunocompetent cells in the central nervous system (CNS). They can be activated by diverse stimuli or factors (Franco and Fernandez-Suarez 2015). A controlled activation of microglia can be beneficial. However, an exacerbated or prolonged activation of microglia may induce neuronal damage by overproduction of neuroinflammatory mediums such as RANTES, TGF-β1, IL-6, TNF-α, and NO (Bellezza et al. 2013; Louboutin and Strayer 2013; Lund et al. 2006; von Bernhardi 2007). The activated microglia also migrate toward infected site or damaged areas and further promote neuroinflammation (de Haas et al. 2008). The hyperactivated microglial cells eventually precipitate neurodegenerative diseases such as multiple sclerosis, Parkinson’s disease, and Alzheimer’s disease (Cherry et al. 2014). Thus, uses of novel pharmacological agents that control microglial overactivation and related neurotoxic may be an efficacious strategy to curtail neuroinflammation-mediated neurodegenerative diseases (Hurley and Tizabi 2013).
Toll-like receptor 4 (TLR4), abundantly expressed on various cells and especially on microglia, is critically involved in mediating neurological dysfunction and neurodegeneration (Drouin-Ouellet and Cicchetti 2012; Okun et al. 2009). The stimulation of TLR4 by LPS, a potent stimulus for microglial activation, triggers the association of IRAK1 and TRAF6 (Akira and Takeda 2004; Verstak et al. 2009), which form a complex with TAK1 and subsequently activate TAK1 (Akira and Takeda 2004). The activated TAK1 regulates transcription factors such as activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) activation to induce neuroinflammation-associated gene expression (Akira and Takeda 2004; Okun et al. 2009). Thus, blocking the signaling pathway may have great potential in the prevention and treatment of neuroinflammation associaated diseaases.
Small molecules from botanical drugs are a crucial source of biologically active compounds. Pristimerin (20α-3-hydroxy-2-oxo-24-nor-friedela-1-10,3,5,7-tetraen-carboxylic acid-29-methylester), a natural quinonoid triterpene, was extracted from Celastraceae or Hippocrateaceae. Pristimerin displays a milieu of properties such as anti-fungal (Luo et al. 2005), antioxidant (Dos Santos et al. 2010; Hui et al. 2014), anticancer (Wu et al. 2005; Yousef et al. 2016b), and antibacterial (Lopez et al. 2011). It has also been reported to reduce NO production via inhibiting inducible nitric oxide synthase in murine macrophages (Dirsch et al. 1997). Our previous results indicated that pristimerin possesses a potent anti-inflammatory property in vivo (Hui et al. 2003). Recently, we showed that pristimerin significantly inhibited LPS-induced cytokines such as TNF-α and IL-8 production in THP-1 cells (Hui et al. 2014). However, it remains unclear whether pristimerin can directly modulate neuroinflammation in microglial cells. The microglial cell line BV-2 is an effective substitute for primary microglia to study the potential role of active compounds in neuroinflammation. (Henn et al. 2009). We therefore investigated the potential action of pristimerin to curtail LPS-induced neuroinflammation in BV-2 microglia. We further analyzed the neuroprotection of pristimerin against BV-2 microglia-mediated neurotoxicity in a co-culture model. To further investigate the underlying mechanism, the involvement of TRAF6, IRAK1, TAK1, NF-κB, and AP-1 was also examined. In the present study, we tested the hypothesis that pristimerin inhibits LPS-stimulated neuroinflammation in BV-2 microglia cells through modulating IRAK1/TRAF6/TAK1-mediated NF-κB and AP-1 signaling pathways in vitro.
Material and Methods
Reagents
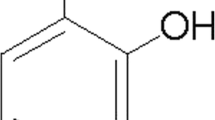
Pristimerin (purity > 98%; Fig. 1a) was purchased from Paypaytech Inc. (Shenzhen, Guangdong, China). It was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) and stored at 4 °C. Enzyme-linked immunosorbent assay (ELISA) kits for mouse TNF-α, TGF-β1, IL-6, and RANTES were obtained from Boster Bio-engineering Co., Ltd. (Wuhan, Hubei, China). Tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), SP600125, pyrrolidine dithiocarbamate (PDTC), LPS from Escherichia coli O127:B8, and protein A/G beads were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Antibodies used for phosphor-c-Jun NH2-terminal protein kinase (p-JNK), JNK, phosphor-mitogen-activated protein kinase kinase 7 (p-MKK7), MKK7, phospho-TAK1 (p-TAK1), TAK1, phospho-IKKα/β (p-IKKα/β), IKKα/β, NF-κB p65, IκB-α, IRAK1, β-actin, and Histone H4 (H4) were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Anti-rabbit IgG-horseradish peroxidase (HRP) and anti-mouse TRAF6 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). RNAiso Plus, Moloney murine leukemia virus (MMLV) reverse transcriptase, Oligo d (T) 18 primers, and SYBR® Premix Ex Taq™ II were obtained from Takara Biotechnology Co., Ltd. (Dalian, Liaoning, China). Biotin-labeled electrophoretic mobility shift assay (EMSA) kits for AP-1 or NF-κB were purchased from Viagene Biotech (Ningbo, Zhejiang, China). A chemiluminescent western blot immunodetection kit was obtained from Pierce SuperSignal (Rockford, IL, USA). Cell lysis buffer for western blot analysis, nuclear protein extraction kit, bicinchoninic acid (BCA) protein assay kit, and NO detecting kit were obtained from the Beyotime Institute of Biotechnology (Jiangsu, China). Other chemicals and reagents were of the highest quality available.

Pristimerin inhibits LPS-induced BV-2 microglial cell death and dampens NO, RANTES, TGF-β1, IL-6, and TNF-α production in LPS-stimulated BV-2 cells. a The chemical structure of pristimerin. b, c Effects of pristimerin on cell viability. BV-2 microglia cells were pretreated with pristimerin (0.15, 0.3, or 0.6 μmol/L) for 3 h without (b) or with LPS (1 μg/mL) for 20 h (c). Cell viability was assayed by the MTT assay. d Representative images of BV2 microglia (scale bar, 200 μm). e–i Pristimerin inhibits LPS-induced productions of NO (e), TNF-α (f), IL-6 (g), RANTES (h), and TGF-β1 (i) in BV-2. BV-2 cells were pretreated with various concentrations of pristimerin for 3 h followed incubation with LPS (1 μg/mL) for 24 h. NO production in the medium was detected by the Griess reagent. The production of TNF-α, IL-6, RANTES, and TGF-β1 in the supernatant was analyzed by ELISA. Results showed as mean ± SEM (n = 3). # P < 0.05, ## P < 0.01, ### P < 0.001 vs. control group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group
Cell Culture
Mouse microglia cell line BV-2 was purchased from the Institute of Basic Medical Sciences of the Chinese Academy of Medical Sciences (Beijing, China). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 μg/mL), and 2 mmol/L l-glutamine at 37 °C in a 5% CO2 incubator. PC12 cells were cultured in DMEM supplemented with FBS (10%), donor horse serum (5%), penicillin (100 U/mL), and streptomycin (100 μg/mL).
MTT Assay
MTT reduction assay was used to quantify cell viability (Sargent and Taylor 1989). BV-2 cells (1 × 104 cells/well) were cultured in 96-well plates. The cells were incubated with differing concentration of pristimerin (0, 0.15, 0.3, or 0.6 μmol/L) for 3 h followed with or without LPS (1 μg/mL) for 20 h. Then, 20 μL of the MTT (5 mg/mL) solution was added to each well, and the cells were incubated for 4 h. Next, 100 mL of 10% SDS in HCl was added. The absorbance of each well was recorded at 570 nm in a Bio-Rad Model 630 microplate reader (Bio-Rad Laboratories, Richmond, CA, USA).
NO Quantification
The accumulation of NO2 −, a stable end-product commonly used as an indicator of NO production, was measured using the Griess reagent. BV-2 cells (3 × 105 cells/well) were seeded in 24-well plates. The cells were treated with different concentrations of pristimerin (0.15, 0.3, or 0.6 μmol/L) for 3 h followed by stimulating with LPS (1 μg/mL) for 24 h. NO production was monitored by measuring the nitrite level accumulated in the culture medium using the NO detecting kit following the manufacturer’s instructions. Optical density was read at 570 nm by a microplate reader (Bio-Rad Model 630 microplate reader).
ELISA Assay
BV-2 cells (3 × 105 cells/well) were preincubated with pristimerin for 3 h, followed by LPS (1 μg/mL) for 24 h in 24-well plates. The culture medium or cell lysates were collected and centrifuged, and the supernatants were aliquoted and stored at − 80 °C until use. The levels of RANTES, TGF-β1, IL-6, and TNF-α were determined by ELISA according to the manufacturer’s instructions. A standard curve was generated during each assay using the mouse RANTES, TGF-β1, IL-6, and TNF-α standards (concentration range 0–1000 pg/mL) provided in the kits. The minimum detectable concentrations of RANTES, TGF-β1, IL-6, and TNF-α were 15.6, 12.5, 15.6, and 15.6 pg/mL, respectively.
qPCR
BV-2 cells (1 × 106 cells/well) were seeded in 6-well plates overnight. The cells were preincubated with pristimerin for 3 h, followed by LPS (1 μg/mL) for 6 h. Total RNA was isolated from BV-2 cells using the RNAiso Plus reagent according to the manufacturer’s protocol. The RNA purity and concentration were assessed with the BioPhotometer (Eppendorf, Germany). The extracted RNA was dried, dissolved in RNase-free water, and stored at − 80 °C before use. Total RNA (1.0 μg) was reverse-transcribed to complementary DNA (cDNA) using M-MLV reverse transcriptase (Takara). Real-time PCR was carried out on the Mastercycler® ep realplex (Eppendorf) using a fluorescent dye, SYBR® Premix Ex Taq™ II (Takara). The primers of all genes used for real-time PCR are shown in Table 1. PCR reactions were performed in triplicate with initial denaturation at 95 °C for 2 min, followed by 40 cycles of amplification at 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s. The housekeeping gene, GAPDH, or β-actin was also amplified in each run as an internal control to normalize the expression of each gene. The relative quantity of cDNA copies was given as a ratio of the target gene to the housekeeping reference gene using the delta-delta Ct method. Melting curves of the amplified DNA products were routinely performed to determine the specificity of the PCR reaction.
Cell Migration Assay
The Costar® transwell system (8-μm pore size polycarbonate membrane) (Costar, USA) was used to evaluate cell migration (Kim et al. 2014). BV-2 cells (2 × 105) in serum-free medium (0.4 mL) were added to the upper well, and serum-free medium (0.4 mL) was added to the lower chamber. Before performing the migration assay, cells were pretreated with pristimerin (0.15, 0.3, 0.6 μmol/L) or solvent control for 3 h. Then, an addition of LPS (1 μg/mL) or solvent control was made to the culture medium in the lower chamber. At the end of an 8-h incubation period, cells that did not migrate and remained on the upper surface of the filter were carefully removed. The migrating cells were stained with crystal violet and quantified via microscopy. The stained cells were counted as the mean number of cells per nine random fields for each assay. All treatments were performed in triplicate.
Neurotoxicity of Activated BV-2 Microglia-Conditioned Medium on PC12 Cells
PC12 cells (2 × 104) were plated in 96-well plates and allowed to settle for 24 h before replacement with conditioned media. To prepare BV-2 conditioned media (CM), BV-2 cells were cultured in a 24-well plate at a density of 3 × 105 cells/well. The conditioned media groups were setup as follows (Cui et al. 2015; Cui et al. 2012): (1) the conditioned media (CM) from control BV-2 cells (Control-CM); (2) LPS (1 μg/mL) was added to the conditioned media from control BV-2 cells (Control-CM + LPS); (3) pristimerin (0.6 μmol/L) alone was added to the conditioned media from control BV-2 cells (Control-CM + Pris); (4) the conditioned media from LPS alone-treated BV-2 cells (LPS-CM); (5) the conditioned media from LPS/pristimerin-treated BV-2 cells (LPS-Pris-CM);(6) 50 μmol/L PDTC (inhibitor of p65 NF-κB) alone was added to the conditioned media from control BV-2 cells (Control-CM + PDTC); (7) 10 μmol/L SP600125 (inhibitor of JNK/AP-1) alone was added to the conditioned media from control BV-2 cells (Control-CM + SP); (8) the conditioned media from LPS/PDTC (50 μmol/L)-treated BV-2 cells (LPS-PDTC-CM); and (9) the conditioned media from LPS/SP600125 (10 μmol/L)-treated BV-2 cells (LPS-SP-CM); briefly describing, BV-2 microglia were pretreated with pristimerin (for 3 h), PDTC (for 30 min), SP600125 (for 30 min), and then stimulated by LPS for 24 h. Then, the media were transferred into PC12 cells. And PC12 cell viability was assessed by MTT assay after 24-h incubation. Changes in the morphology of PC12 cells in various groups were observed using a phase-contrast microscope.
Western Blot Analysis
The western blot analysis was performed as described previously (Hui et al. 2008). Briefly, the cell extracts were run on SDS-polyacrylamide gels (10%) and then transferred onto nitrocellulose membranes. After 1 h of blocking in 5% skim milk, the membranes were incubated with primary antibodies against phosphorylated or total forms of JNK (1:2000), MKK7 (1:1000), TAK1 (1:2000), IKKα/β (1:1000), NF-κB p65 (1:500), IκB-α (1:1000), TRAF6 (1:1000), IRAK1 (1:1000), β-actin (1:2000), and Histone H4 (1:1000) overnight at 4 °C. After the membranes were washed three times with TBST (Tris-buffered saline containing Tween) and then incubated with anti-mouse IgG-HRP or goat anti-rabbit IgG-HRP for 1 h, the intensity of the specific bands was detected using the chemiluminescent western blot immunodetection kit. The optical densities of the bands were analyzed using Image-Pro plus 5.1 (Media Cybernetics, San Diego, CA, USA). Histone H4 or actin was used as the loading control for the total protein.
Electrophoretic Mobility Shift Assay
Nuclear proteins were extracted and isolated using the nuclear protein extraction kit. The nuclear pellets were suspended in nuclear extraction buffer provided by the manufacturer and incubated on ice for 30 min with occasional gentle shaking. After centrifugation at 12,000×g for 10 min, the supernatant was harvested as a nuclear protein and stored at − 80 °C until use. Protein concentrations were determined using the BCA protein assay kit.
The EMSA was used to study the effect of pristimerin on NF-κB or AP-1 binding following the manufacturer’s (Viagene Biotech) protocol. Briefly, nuclear extracted proteins were incubated with biotin-labeled, double-stranded NF-κB or AP-1 consensus oligonucleotide probe at room temperature for 20 min. The specificity of binding of polypeptide factors to NF-κB or AP-1 was carried out using excess of unlabeled competitive NF-κB or AP-1 probes (cold oligonucleotides), respectively. The reaction mixture was analyzed by electrophoresis in native polyacrylamide gels, transferred to nylon membranes, and subject to UV cross-linking. After the membrane was blocked, streptavidin-horseradish peroxidase conjugated solution was added and biotinylated DNA was detected using Lighten® HRP-B Substrate Solution supplied by the manufacturer (Viagene Biotech).
Immunoprecipitation Assay
BV-2 cells were preincubated with pristimerin (0.6 μmol/L) for 3 h and then stimulated with LPS for 30 min. The cells were rinsed three times with ice-cold phosphate buffered saline and then lysed in lysis buffer (50 mM of Tris-HCl, 1 mM of EDTA, 150 mmol/L of NaCl, 10 mmol/L of NaF, 0.5% NP-40, 10% glycerol, 1% Triton X-100, 1 mmol/L of PMSF, and 1 mmol/L of protein inhibitor cocktail). Total cell lysates were preincubated with anti-TRAF6 antibody for 1 h and then incubated with protein A/G beads overnight. Immunocomplexes were recovered by washing the resin 4 times and re-suspended in SDS-sample buffer. The immunoprecipitates were subjected to SDS-PAGE and immunoblotting with specific primary antibodies.
Statistical Analysis
Data are expressed as the means ± SEM. Data from three independent experiments were analyzed by one-way analysis of variance (ANOVA) followed by the Dunnett’s t test. (SPSS 17.0, software, USA). A P value below 0.05 was considered to be statistically significant.
Results
Pristimerin Inhibits LPS-Induced BV-2 Microglial Cell Death and Dampens NO, RANTES, TGF-β1, IL-6, and TNF-α Production in LPS-Stimulated BV-2 Cells
Prior to studying the effect of pristimerin (Fig. 1a) on BV-2 microglia activation, we determined the potential cytotoxic action of pristimerin on BV-2 microglial cells using the MTT assay. As shown in Fig. 1b, d, 0.15–0.6 μmol/L pristimerin did not exert toxic effects to BV-2 cell. Therefore, 0.15–0.6 μmol/L pristimerin were chosen for the subsequent experiments. As shown in Fig. 1c, d, 1 μg/mL LPS decreased BV-2 cell viability as compared to vehicles groups (P < 0.001); however, pristimerin (0.3, and 0.6 μmol/L) rescued BV-2 microglial cells from LPS-induced toxicity (P < 0.05). We next measured the effect of pristimerin on the production of NO. Fig. 1e shows that LPS (1 μg/mL) induced a remarkable production of NO (p < 0.001). However, BV-2 cell pretreatment with pristimerin (0.15–0.6 μmol/L) markedly reduced LPS-induced NO production. Proinflammatory cytokines such as TGF-β1, IL-6, TNF-α, and chemokines such as RANTES are also critical mediators of neuroinflammation. Therefore, we analyzed the effects of pristimerin on the production of these inflammatory mediators in LPS-stimulated BV-2 microglia cells. The concentration of TNF-α increased about 23.7-fold in LPS-stimulated BV-2 cells as compared with control (Fig. 1f). Pristimerin at doses of 0.15, 0.3, and 0.6 μmol/L markedly decreased TNF-α concentrations to 86.0 ± 5.4, 68.3 ± 7.5, and 64.2 ± 3.9%, respectively (Fig. 1f). Similarly, LPS-induced IL-6 (Fig. 1g) and RANTES (Fig. 1h) and TGF-β1 (Fig. 1i) productions were notably reduced by pristimerin. In all experiments, pristimerin alone had no influence on NO, TNF-α, IL-6, TGF-β1, and RANTES production in non-stimulated BV-2 cells (data not shown).
Pristimerin Suppresses mRNA Expression of RANTES, IL-6, TGF-β1, and TNF-α
To investigate the mechanisms involved in the suppressive effects of pristimerin on the production of RANTES, IL-6, TGF-β1, and TNF-α, we used qPCR to determine the messenger RNA (mRNA) levels of inflammatory mediators. As shown in Fig. 2a–d, the mRNA expression of TNF-α, IL-6, TGF-β1, and RANTES, markedly upregulated by LPS, was blocked by pristimerin (0.15–0.6 μmol/L). The results indicate that pristimerin negatively controls the expression of RANTES, TNF-α, IL-6, and TGF-β1 at the transcriptional level.

Pristimerin decreased the mRNA expressions of TNF-α, IL-6, TGF-β1, and RANTES induced by LPS in BV-2 cells. BV-2 microglia were incubated with pristimerin (0.15, 0.3, or 0.6 μmol/L) for 3 h and then stimulated with LPS (1 μg/mL) for 6 h. Total RNA was isolated after LPS treatment; the mRNA levels of TNF-α (a), IL-6 (b), TGF-β1 (c), and RANTES (d) were determined by quantitative real-time PCR. Results showed as mean ± SEM (n = 3). ## P < 0.01 vs. control group, ### P < 0.001 vs. control group; *P < 0.05, **P < 0.01 vs. LPS group
Pristimerin Inhibits Migration of BV-2 Microglia In Vitro
Since pristimerin suppresses the production of various proinflammatory mediators, we next investigated whether pristimerin inhibits the motility of BV-2 microglia. The ability of pristimerin to block motility of microglia was assessed via the transwell migration assay in vitro. As shown in Fig. 3a, b, LPS markedly increased the migratory potential of the BV-2 cells as compared with the control groups, while pristimerin significantly blocked the migration of BV-2 cell induced by LPS. Furthermore, BV-2 cell migration was not significantly affected by pristimerin alone as compared with the control groups (Fig. 3a, b). This result suggests that pristimerin blocks LPS-induced migration of BV-2 microglia.

Pristimerin inhibited migration of BV-2 microglia cell in vitro. Transwell migration assay shown that pristimerin reduced the migration of BV-2 microglia. a Light microscopy images of BV-2 cells treated with pristimerin of different concentrations or the control were shown (scale bar, 50 μm). b The quantitative analysis reveals a decrease in the migration of microglia with pristimerin treatment. Data are represented as mean ± SEM (n = 3), ### P < 0.001 vs. control group; *P < 0.05, ***P < 0.001 vs. LPS group
Pristimerin Ameliorates Microglia-Mediated Neuronal Cell Death in Conditioned Media System
Activated microglia release many neuroinflammatory mediators that can impair surrounding neurons. Since pristimerin suppresses the release of some of these neuroinflammatory mediators, we asked whether pristimerin might protect neuronal cells from BV-2 microglia-induced neurotoxicity (He et al. 2014; Westerink and Ewing 2008). The neurotoxicity of activated BV-2 conditioned media to PC 12 neuronal cells was assayed by the MTT assay. The data shown that the Control-CM + LPS or Control-CM + Pris did not affect PC12 cell viabilities (Fig. 4a), whereas LPS-CM markedly reduced PC12 cell viability (P < 0.01; Fig. 4a). The decreased of PC12 neuronal cells viability was reversed by LPS-Pris-CM (P < 0.01; Fig. 4a). Microscopic images provided morphological evidence to confirm MTT results (Fig. 4b). To explore whether pristimerin ameliorates BV-2 microglia-mediated PC12 cell death was involved with NF-κB and JNK/AP-1 cell pathways, a specific p65 NF-κB inhibitor PDTC and JNK/AP-1 inhibitor SP600125 were used. It is well known that PDTC and SP600125 inhibit inflammatory mediator’s production in LPS-activated microglial cells via NF-κB and JNK/AP-1 cell signal, respectively. (Ma et al. 2017; Waetzig et al. 2005; Yoshioka et al. 2010). As shown in Fig. 4a, PDTC or SP600125 pretreatment of BV-2 microglia markedly decreased the cell death of PC-12 cells after LPS-CM stimulation in conditioned media system. However, PDTC or SP600125 themselves affected neither the survival PC12 cells nor the morphology as analyzed by MTT assays (Fig. 4a, b). These results indicate that pristimerin protects PC12 neuronal cells against overactivated BV-2 microglia cell-mediated neurotoxicity. NF-κB and JNK/AP-1 cell pathways might be involved in the anti-neurotoxicity effects of pristimerin.

Pristimerin protected PC12 cells from death induced by LPS-activated BV-2 cell-mediated conditioned media. a Cell viabilities of PC12 cells were measured using MTT assay. b Light microscopy images of PC-12 cells (scale bar, 50 μm). Groups are summarized as follows: Control-CM, Control-CM + LPS, Control-CM + pristimerin, Control-CM + PDTC, Control-CM + SP, LPS-PDTC-CM, LPS-SP-CM LPS-CM, LPS-Prist-CM. The data were replicated in three repeated independent experiments. Results showed as mean ± SEM. ## P < 0.01 vs. Control-CM group; *P < 0.05 vs. LPS-CM group, **P < 0.01 vs. LPS-CM group
Pristimerin Blocks NF-κB DNA Binding Activity in LPS-Activated BV-2 Microglia
To evaluate the inhibition of pristimerin on the NF-κB pathway, EMSA was used to estimate the NF-κB DNA-binding activity. The nuclear extracts from BV-2 cells showed strong DNA binding ability of NF-κB following LPS stimulation (Fig. 5a, line 2; Fig. 5b). This activity was markedly inhibited after pristimerin treatment (Fig. 5a, lanes 4, 5, and 6; Fig. 5b). To determine whether the shifted band was specific for NF-κB, competition test using an excess of unlabeled probe was performed. As shown in Fig. 5a (lane 3) and Fig. 5b, excessive amounts of unlabeled probe completely reversed the LPS-induced bound complex. These data showed that pristimerin blocks NF-κB DNA binding activity in LPS-activated BV-2 microglia.

Pristimerin blocked LPS-induced NF-κB-DNA binding activity, IKKα/β phosphorylation, IκB-α degradation, and p65-NF-κB nuclear translocation in LPS-stimulated BV-2 cells. BV-2 cells were pretreated with 0.15–0.6 μmol/L pristimerin for 3 h then stimulated with 1 μg/mL LPS for 30 min. a, b Nuclear protein was isolated and analyzed for NF-κB binding by EMSA. Total proteins were prepared, and western blot analysis was performed using specific p-IKKα/β (c) or IκB-α antibody (d). β-Actin or IKKα/β was used as a control for protein loading and integrity. Cytosolic (e) and nuclear (f) extracts were isolated, and the levels of p65 NF-κB in each fraction were determined by western blot analysis. Histone H4 and β-actin were used as internal controls. The data shown are representative of three independent experiments. Results showed as mean ± SEM. ### P < 0.001 vs. control group; **P < 0.01, ***P < 0.001 vs. LPS group
Pristimerin Inhibits IKKα/β Phosphorylation, IκBα Degradation, and Nuclear Translocation of NF-κB in LPS-Activated BV-2 Microglia
LPS induces IKK complex, leading to the phosphorylation and degradation of IκB-α and nuclear translocation of NF-κB. Therefore, immunoblotting was performed to examine the effects of pristimerin on LPS-induced IKKα/β activation, IκBα degradation, and nuclear translocation of NF-κB. As shown in Fig. 5, LPS stimulation remarkably promoted the phosphorylation of IKKα/β and correspondingly decreased the expression of IκB-α protein (Fig. 5c, d). Pristimerin markedly blocked LPS-induced phosphorylation of IKKα/β as well as degradation of IκB-α (Fig. 5c, d). In addition, pristimerin significantly suppressed LPS-induced nuclear translocation of NF-κB p65 (Fig. 5e, f).
Pristimerin Blocks DNA Binding Activity of AP-1 in LPS-Stimulated BV-2 Microglia
AP-1 is also a central transcription factor in gene expression (Guha and Mackman 2001). The effect of pristimerin on DNA binding activity of AP-1 was performed by EMSA. As shown in Fig. 6, an elevation in DNA and nuclear protein complexes was detected in LPS-activated BV-2 cells by using the probe of AP-1 (Fig. 6, lane 2). Such elevation in DNA and protein complex was markedly inhibited by pristimerin preconditioned (Fig. 6, lanes 4–6). Furthermore, the DNA and protein complex was completely inhibited by an excess of unlabeled AP-1 oligonucleotide (Fig. 6, lane 3).

Pristimerin arrested LPS-induced AP-1-DNA binding activity in LPS-stimulated BV-2 cells. Nuclear extracts were prepared and analyzed for the DNA binding activity of AP-1 by EMSA. Binding specificity was determined using the excess unlabeled probe (referred to as “cold”) to compete with the labeled oligonucleotide. BV-2 microglia were pretreated with pristimerin (0.15, 0.3, or 0.6 μmol/L) for 3 h before stimulation with LPS (1 μg/mL) for another 30 min. The results shown are representative of three independent experiments. ### P < 0.001 vs. control group (cultured in the medium alone); **P < 0.01, ***P < 0.001 vs. LPS group
Pristimerin Inhibits MKK7/JNK Activation in LPS-Stimulated BV-2 Microglia
MKK7 activates the JNK pathway that regulates inflammatory and stress responses (Davis 2000; Kragelj et al. 2015). To elucidate the mechanism underlying the protective effect of pristimerin on neuroinflammation in BV-2 microglia, the protein expression levels of the JNK and MKK7 were assayed. The results revealed that MKK7 and JNK were phosphorylated by the stimulation with LPS (Fig. 7a, b). Pretreatment of BV-2 cells with pristimerin (0.15, 0.3 and 0.6 μmol/L) markedly suppressed LPS-induced phosphorylation of JNK and MKK7 (P < 0.05) (Fig. 7a, b).

Pristimerin inhibited the phosphorylation of JNK and MKK7. BV-2 microglia were pretreated with pristimerin (0.15, 0.3, or 0.6 μmol/L) for 3 h and then stimulated with LPS (1 μg/mL) for 30 min. Cell lysates were prepared and analyzed for JNK (Fig 7a) or MKK7 (Fig 7b) by western blotting. The data were confirmed by three independent experiments with freshly prepared reagents. # P < 0.05, ### P < 0.001 vs. control group; **P < 0.01, ***P < 0.001 vs. LPS group
Pristimerin Arrests IRAK1 Association to TRAF6 and Limits TAK1 in Activated BV-2 Microglia
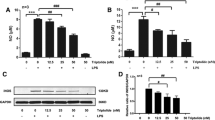
The association of TRAF6 with IRAK1 ultimately activated TAK1. The activated TAK1 bifurcated signal into two important branches, the MKK7/JNK pathway and the IKK-dependent cascade. To determine the role of TRAF6, IRAK1, and TAK1 signalosome formation in the action of pristimerin on LPS-induced activation of BV-2 microglia, we determined the TRAF6 binds IRAK1 using co-immunoprecipitation assays, the activation of TAK1 using Western blotting (Fig. 8). The results shown that LPS markedly promoted the expression of TRAF6 (Fig. 8a), increased degradation of IRAK1 (Fig.8b), facilitated the formation of the TRAF6-IRAK1 complex, and enhanced the phosphorylation of TAK1 (Fig. 8c). Pretreatment with pristimerin markedly suppressed the LPS-induced expression of TRAF6, degradation of IRAK1, formation of the TRAF6-IRAK1 complex and phosphorylation of TAK1 (Fig. 8a–d).

Pristimerin inhibits LPS-induced expression of TRAF6, IRAK1, and p-TAK1 and arrests the interactions of IRAK1 with TRAF6 in BV-2 microglia. BV-2 cells were pretreated with 0.15, 0.3, or 0.6 μmol/L of pristimerin for 3 h and stimulated with LPS (1 μg/mL) for 30 min. The protein levels of TRAF6 (a), IRAK1 (b), and p-TAK1 (d) in the whole cell lysate were analyzed by western blotting. c The TRAF6-IRAK1 complex was first precipitated by an antibody against TRAF6 and then analyzed by western blotting for IRAK1 and TRAF6. ## P < 0.01, ### P < 0.001 vs. control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group
Discussion
Overactivated microglia play a core role in the pathogenesis of neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (Cherry et al. 2014). Activated microglia trigger neuroinflammation and induce neuronal death by excessive production of NO and proinflammatory cytokines and chemokines such as TNF-α, TGF-β1, IL-6, and RANTES (Helmy et al. 2011; Louboutin and Strayer 2013; Lund et al. 2006; Rubio-Perez and Morillas-Ruiz 2012; Ziebell and Morganti-Kossmann 2010). Furthermore, the inflammatory mediators exacerbate blood-brain barrier disruption, enable the entry of leukocytes into the brain, and attract microglia in promoting inflammatory responses in CNS (Laflamme and Rivest 1999). It has been well demonstrated that activation of TLR4 is intimately involved in signaling of NF-κB and AP-1 (Okun et al. 2009). Inappropriate activation of NF-κB and AP-1 leads to overproduction of inflammatory mediators, which play key roles in neuroinflammation (Li and Verma 2002; Nam 2006; Okun et al. 2009). In this signal transduction cascade, IRAK1 is activated and sequentially forms complex with TRAF6, which mediates activation of TAK1 (Brown et al. 2011). Phosphorylation of TAK1 in turn activates IKKα/β, which leads to IκB-α degradation, and allows for NF-κB to translocate to the nucleus and initiates NF-κB-dependent transcription (Brown et al. 2011). Concurrently, TAK1 phosphorylates MKK7/JNK subsequent AP-1 activation (Gatheral et al. 2012; Gingery et al. 2008; Wang et al. 2001) (Fig. 9). Therefore, inhibition of the abnormal activation of microglia by pharmacological agents may be a potential therapeutic strategy to curtail neurodegenerative diseases.

Schematic model of pristimerin on LPS-triggered neurotoxicity in BV-2 microglia cells through modulating IRAK1/TRAF6/TAK1 mediated NF-κB and AP-1 signaling pathways in vitro
Natural products are potential sources of anti-neuroinflammatory drugs, such as celastrol, curcumin, baicalein, berberine, luteolin, and resveratrol, which have been reported to suppress LPS-activated microglial cells via NF-κB pathway (Chen et al. 2004; Cianciulli et al. 2016; Kang et al. 2004; Kim et al. 2006; Suk et al. 2003). Especially, celastrol, a chemical structure similar to pristimerin, is a potent anti-inflammatory and antioxidant triterpene from Celastraceae family. It blocks microglial activation via inhibiting NO, proinflammatory cytokines, and chemokines production (He et al. 1998; Jung et al. 2007; Nakamichi et al. 2010). Recent studies, including those of our own, demonstrated that pristimerin possess significantly anti-inflammatory, anticancer, and anti-tumor angiogenesis effects (Deng et al. 2015; Yousef et al. 2016a). Pristimerin also reduces the expression of proangiogenic factors including TNF-α, Ang-1, and MMP-9 in sera of inflamed joints (Deng et al. 2015; Yousef et al. 2016a). However, whether or not pristimerin exerts a protective action on LPS-triggered neurotoxicity in BV-2 microglia is less well described. The aim of this study was to evaluate the potential effect of pristimerin on LPS-induced neurotoxicity in BV-2 microglia cells in vitro. Although the LPS-triggered microglia secretion is not the same as in vivo conditions, it could mimic the pathological conditions in neurodegenerative diseases where activated microglia contributes to neuronal damage (Henn et al. 2009). In the present study, 1 μg/mL LPS was used to activate BV-2 microglia cells. The concentration of LPS used and the time stimulated depended on our preliminary experiments and related literatures (Lee et al. 2012; Zeng et al. 2015). The present results showed that LPS decreased BV-2 cell survival. LPS alone also markedly induced microglia cells to produce TNF-α, TGF-β1, IL-6, RANTES, NO, and augmented migration of BV-2 microglia. Notably, the conditioned medium from LPS-challenged BV-2 cells markedly caused PC12 neuronal death. PDTC, an inhibitor of p65 NF-κB, and SP600125, an inhibitor of JNK/AP-1, also significantly decreased PC12 neuronal death induced by LPS-challenged BV-2 cells. Moreover, LPS alone significantly activated IRAK1-TRAF6-TAK1 cell signal pathway and concomitantly triggered the activation of nucleus transcription factor such as AP-1 and NF-κB. The results indicated that LPS activates transcripts such as AP-1 and NF-κB, which are regulated by IRAK1-TRAF6-TAK1 signal pathway, resulting in the overproduction of inflammatory mediators such as TNF-α, TGF-β1, IL-6, RANTES, NO, and subsequent neuronal damage. The in vitro study also indicated a potential correlation between overproduction of neuroinflammatory mediums by hyperactivated microglial cells and neuronal damage.
In the present study, pristimerin alone did not affect the viability of BV-2 cells; however, pristimerin has a protective role against LPS-induced microglial cell death. Pristimerin is also a potent inhibitor of the LPS-induced neuroinflammatory molecules such as RANTES, TGF-β1, TNF-α, IL-6, and NO in BV-2 microglia. Similarly, pristimerin inhibited LPS-induced NO, TNF-α, and IL-6 production in murine macrophages RAW264.7 or human monocytic THP-1 cells (Dirsch et al. 1997; Hui et al. 2014; Kim et al. 2013). These results, together with our present study, strongly indicate that pristimerin is a potent inhibitor of inflammation and neuroinflammation.
The activation of microglia is characterized by microglial cell migration. The microglial cell migration also promotes the neuroinflammatory response. The present study showed that pristimerin significantly inhibited LPS induced BV-2 microglial cell migration, which is consistent with the study that pristimerin markedly inhibited VEGF-induced HUVEC migration (Deng et al. 2015). The blockade of microglial cell migration by pristimerin may be ascribing to the inhibition the production of proinflammatory cytokines and chemokine. Furthermore, the reduced microglial cell migration by pristimerin, in turn, inhibits hyperactivated microglial cells triggered neurotoxicity.
Neuron-like PC12 cell model is widely used as an in vitro neuronal model (Westerink and Ewing 2008). In the present study, we used PC12 cells as a neuronal model system to examine the protective effects of pristimerin on LPS-activated microglia toxicity. It was confirmed that pristimerin exerted protective effects against activated BV-2 cell-mediated neuronal cell injury, thus strengthening the notion that pristimerin inhibits microglial neurotoxicity via suppressing the production of inflammatory molecules.
Among the subunits of NF-κB, the p65 is the most prevalent activated form of NF-κB. Moreover, P65 NF-κB is also a classical pathway. Indeed, pristimerin has been reported to inhibit LPS-induced NF-κB activation in murine macrophage RAW264.7 cells and human monocytic THP-1 cells (Hui et al. 2014; Kim et al. 2013). Nevertheless, little has been known about the molecular action of pristimerin on upstream of p65 NF-κB activation in LPS-induced microglia. Furthermore, it is not known whether pristimerin inhibits AP-1 activity in LPS-induced microglia. To further elucidate the anti-neurotoxicity mechanism of pristimerin, we investigated IRAK1-TRAF6-TAK1 pathway, which regulates NF-κB/AP-1-mediated inflammatory molecules production in LPS-activated microglia. Our results showed that pristimerin evidently inhibited TRAF6 expression and IRAK1 degradation. Moreover, pristimerin significantly blocked the formation of the complex of IRAK1 with TRAF6, limited TAK1 activation, and subsequently suppressed the IKKα/β/NF-κB and MKK7/JNK/AP-1 pathways in LPS-stimulated BV-2 microglia cells. Meanwhile, treatment of the cells with pristimerin alone had no influence on the aforementioned effects (data not shown). Interestingly, celastrol also blocks LPS-activated BV-2 microglial cell via inhibition of MAPK phosphorylation and NF-κ B activation (Jung et al. 2007). All these results suggest that the conjugated diosphenol function, a common chemical structure of pristimerin and celastrol, might play a vital role in their inhibitory action on microglial activation (Morita et al. 2008).
Taken together, these results provide insights that pristimerin blocks LPS-induced neurotoxicity via regulating IRAK1-TRAF6-TAK1-mediated NF-κB and AP-1 cell signal pathway in BV-2 microglial cells. However, the neuroprotective actions of pristimerin in vivo need to be explored further.
References
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511. https://doi.org/10.1038/nri1391
Bellezza I et al (2013) Furanodien-6-one from Commiphora erythraea inhibits the NF-kappaB signalling and attenuates LPS-induced neuroinflammation. Mol Immunol 54:347–354. https://doi.org/10.1016/j.molimm.2013.01.003
Brown J, Wang H, Hajishengallis GN, Martin M (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90:417–427. https://doi.org/10.1177/0022034510381264
Chen CJ, Raung SL, Liao SL, Chen SY (2004) Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem Pharmacol 67:957–965
Cherry JD, Olschowka JA, O'Banion MK (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98. https://doi.org/10.1186/1742-2094-11-98
Cianciulli A, Calvello R, Porro C, Trotta T, Salvatore R, Panaro MA (2016) PI3k/Akt signalling pathway plays a crucial role in the anti-inflammatory effects of curcumin in LPS-activated microglia. Int Immunopharmacol 36:282–290. https://doi.org/10.1016/j.intimp.2016.05.007
Cui Y et al (2012) Neuroprotective effect of methyl lucidone against microglia-mediated neurotoxicity. Eur J Pharmacol 690:4–12. https://doi.org/10.1016/j.ejphar.2012.05.041
Cui Y et al (2015) Dieckol attenuates microglia-mediated neuronal cell death via ERK, Akt and NADPH oxidase-mediated pathways. Korean J Physiol Pharmacol 19:219–228. https://doi.org/10.4196/kjpp.2015.19.3.219
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239–252
de Haas AH, Boddeke HW, Biber K (2008) Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 56:888–894. https://doi.org/10.1002/glia.20663
Deng Q, Bai S, Gao W, Tong L (2015) Pristimerin inhibits angiogenesis in adjuvant-induced arthritic rats by suppressing VEGFR2 signaling pathways. Int Immunopharmacol 29:302–313. https://doi.org/10.1016/j.intimp.2015.11.001
Dirsch VM, Kiemer AK, Wagner H, Vollmar AM (1997) The triterpenoid quinonemethide pristimerin inhibits induction of inducible nitric oxide synthase in murine macrophages. Eur J Pharmacol 336:211–217
Dos Santos VA, Dos Santos DP, Castro-Gamboa I, Zanoni MV, Furlan M (2010) Evaluation of antioxidant capacity and synergistic associations of quinonemethide triterpenes and phenolic substances from Maytenus ilicifolia (Celastraceae). Molecules 15:6956–6973. https://doi.org/10.3390/molecules15106956
Drouin-Ouellet J, Cicchetti F (2012) Inflammation and neurodegeneration: the story ‘retolled’. Trends Pharmacol Sci 33:542–551. https://doi.org/10.1016/j.tips.2012.07.002
Franco R, Fernandez-Suarez D (2015) Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 131:65–86. https://doi.org/10.1016/j.pneurobio.2015.05.003
Gatheral T et al (2012) A key role for the endothelium in NOD1 mediated vascular inflammation: comparison to TLR4 responses. PLoS One 7:e42386. https://doi.org/10.1371/journal.pone.0042386
Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ (2008) TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res 314:2725–2738. https://doi.org/10.1016/j.yexcr.2008.06.006
Guha M, Mackman N (2001) LPS induction of gene expression in human monocytes. Cell Signal 13:85–94
He W, Huang FC, Gavai A, Chan WK, Amato G, KT Y, Zilberstein A (1998) Novel cytokine release inhibitors. Part III: truncated analogs of tripterine. Bioorg Med Chem Lett 8:3659–3664
He Q et al (2014) Trehalose alleviates PC12 neuronal death mediated by lipopolysaccharide-stimulated BV-2 cells via inhibiting nuclear transcription factor NF-kappaB and AP-1 activation. Neurotox Res 26:430–439. https://doi.org/10.1007/s12640-014-9487-7
Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ (2011) The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab 31:658–670. https://doi.org/10.1038/jcbfm.2010.142
Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009) The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26:83–94
Hui B,Wu YJ, Wang H, Tian X (2003) Effect of pristimerin on experimental inflammation in mice and rats. Chin Pharm Bull 19:656–659
Hui B, Li J, Geng MY (2008) Sulfated polymannuroguluronate, a novel anti-acquired immune deficiency syndrome drug candidate, decreased vulnerability of PC12 cells to human immunodeficiency virus tat protein through attenuating calcium overload. J Neurosci Res 86:1169–1177. https://doi.org/10.1002/jnr.21566
Hui B, Yao X, Zhou Q, Wu Z, Sheng P, Zhang L (2014) Pristimerin, a natural anti-tumor triterpenoid, inhibits LPS-induced TNF-alpha and IL-8 production through down-regulation of ROS-related classical NF-kappaB pathway in THP-1 cells. Int Immunopharmacol 21:501–508. https://doi.org/10.1016/j.intimp.2014.06.010
Hurley LL, Tizabi Y (2013) Neuroinflammation, neurodegeneration, and depression. Neurotox Res 23:131–144. https://doi.org/10.1007/s12640-012-9348-1
Jung HW, Chung YS, Kim YS, Park YK (2007) Celastrol inhibits production of nitric oxide and proinflammatory cytokines through MAPK signal transduction and NF-kappaB in LPS-stimulated BV-2 microglial cells. Exp Mol Med 39:715–721. https://doi.org/10.1038/emm.2007.78
Kang G, Kong PJ, Yuh YJ, Lim SY, Yim SV, Chun W, Kim SS (2004) Curcumin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression by inhibiting activator protein 1 and nuclear factor kappab bindings in BV2 microglial cells. J Pharmacol Sci 94:325–328
Kim JS, Lee HJ, Lee MH, Kim J, Jin C, Ryu JH (2006) Luteolin inhibits LPS-stimulated inducible nitric oxide synthase expression in BV-2 microglial cells. Planta Med 72:65–68. https://doi.org/10.1055/s-2005-873145
Kim HJ, Park GM, Kim JK (2013) Anti-inflammatory effect of pristimerin on lipopolysaccharide-induced inflammatory responses in murine macrophages. Arch Pharm Res 36:495–500. https://doi.org/10.1007/s12272-013-0054-1
Kim EA, Han AR, Choi J, Ahn JY, Choi SY, Cho SW (2014) Anti-inflammatory mechanisms of N-adamantyl-4-methylthiazol-2-amine in lipopolysaccharide-stimulated BV-2 microglial cells. Int Immunopharmacol 22:73–83. https://doi.org/10.1016/j.intimp.2014.06.022
Kragelj J, Palencia A, Nanao MH, Maurin D, Bouvignies G, Blackledge M, Jensen M (2015) Structure and dynamics of the MKK7–JNK signaling complex. Proc Natl Acad Sci U S A 112:3409–3414. https://doi.org/10.1073/pnas.1419528112
Laflamme N, Rivest S (1999) Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor kappaB alpha within specific cellular populations of the rat brain. J Neurochem 73:309–321
Lee KW, Jung SY, Choi SM, Yang EJ (2012) Effects of ginsenoside Re on LPS-induced inflammatory mediators in BV2 microglial cells. BMC Complement Altern Med 12:196. https://doi.org/10.1186/1472-6882-12-196
Li Q, Verma IM (2002) NF-kappaB regulation in the immune system. Nat Rev Immunol 2:725–734. https://doi.org/10.1038/nri910
Lopez MR, de Leon L, Moujir L (2011) Antibacterial properties of phenolic triterpenoids against Staphylococcus epidermidis. Planta Med 77:726–729. https://doi.org/10.1055/s-0030-1250500
Louboutin JP, Strayer DS (2013) Relationship between the chemokine receptor CCR5 and microglia in neurological disorders: consequences of targeting CCR5 on neuroinflammation, neuronal death and regeneration in a model of epilepsy. CNS Neurol Disord Drug Targets 12:815–829
Lund S et al (2006) The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol 180:71–87. https://doi.org/10.1016/j.jneuroim.2006.07.007
Luo DQ, Wang H, Tian X, Shao HJ, Liu JK (2005) Antifungal properties of pristimerin and celastrol isolated from Celastrus hypoleucus. Pest Manag Sci 61:85–90. https://doi.org/10.1002/ps.953
Ma L, Sun P, Zhang JC, Zhang Q, Yao SL (2017) Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int J Mol Med 40:31–38. https://doi.org/10.3892/ijmm.2017.2987
Morita H, Hirasawa Y, Muto A, Yoshida T, Sekita S, Shirota O (2008) Antimitotic quinoid triterpenes from Maytenus chuchuhuasca. Bioorg Med Chem Lett 18:1050–1052. https://doi.org/10.1016/j.bmcl.2007.12.016
Nakamichi K, Kitani H, Takayama-Ito M, Morimoto K, Kurane I, Saijo M (2010) Celastrol suppresses morphological and transcriptional responses in microglial cells upon stimulation with double-stranded RNA. Int J Neurosci 120:252–257. https://doi.org/10.3109/00207451003615763
Nam NH (2006) Naturally occurring NF-kappaB inhibitors. Mini Rev Med Chem 6:945–951
Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV (2009) Toll-like receptors in neurodegeneration. Brain Res Rev 59:278–292. https://doi.org/10.1016/j.brainresrev.2008.09.001
Rubio-Perez JM, Morillas-Ruiz JM (2012) A review: inflammatory process in Alzheimer's disease, role of cytokines. Scientific World J 2012:756357. https://doi.org/10.1100/2012/756357
Sargent JM, Taylor CG (1989) Appraisal of the MTT assay as a rapid test of chemosensitivity in acute myeloid leukaemia. Br J Cancer 60:206–210
Suk K, Lee H, Kang SS, Cho GJ, Choi WS (2003) Flavonoid baicalein attenuates activation-induced cell death of brain microglia. J Pharmacol Exp Ther 305:638–645. https://doi.org/10.1124/jpet.102.047373
Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A (2009) MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem 284:24192–24203. https://doi.org/10.1074/jbc.M109.023044
von Bernhardi R (2007) Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox Res 12:215–232
Waetzig V et al (2005) c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 50:235–246. https://doi.org/10.1002/glia.20173
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351. https://doi.org/10.1038/35085597
Westerink RH, Ewing AG (2008) The PC12 cell as model for neurosecretion. Acta Physiol (Oxf) 192:273–285. https://doi.org/10.1111/j.1748-1716.2007.01805.x
Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR, YC W (2005) Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer Ther 4:1277–1285. https://doi.org/10.1158/1535-7163.MCT-05-0027
Yoshioka Y, Takeda N, Yamamuro A, Kasai A, Maeda S (2010) Nitric oxide inhibits lipopolysaccharide-induced inducible nitric oxide synthase expression and its own production through the cGMP signaling pathway in murine microglia BV-2 cells. J Pharmacol Sci 113:153–160
Yousef BA, Guerram M, Hassan HM, Hamdi AM, Zhang LY, Jiang ZZ (2016a) Pristimerin demonstrates anticancer potential in colorectal cancer cells by inducing G1 phase arrest and apoptosis and suppressing various pro-survival signaling proteins. Oncol Rep 35:1091–1100. https://doi.org/10.3892/or.2015.4457
Yousef BA, Hassan HM, Zhang LY, Jiang ZZ (2016b) Anticancer potential and molecular targets of pristimerin: a mini-review current cancer drug targets
Zeng KW, Yu Q, Liao LX, Song FJ, Lv HN, Jiang Y, PF T (2015) Anti-neuroinflammatory effect of MC13, a novel coumarin compound from condiment Murraya, through inhibiting lipopolysaccharide-induced TRAF6-TAK1-NF-kappaB, P38/ERK MAPKS and Jak2-Stat1/Stat3 pathways. J Cell Biochem 116:1286–1299. https://doi.org/10.1002/jcb.25084
Ziebell JM, Morganti-Kossmann MC (2010) Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7:22–30. https://doi.org/10.1016/j.nurt.2009.10.016
Acknowledgements
We are grateful to Professor Geng Meiyu (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, China) for kindly providing us with PC12 cells. We also are grateful to Professor Guo Su (Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, CA, USA) for critically editing the paper.
Funding
This study was supported by the Foundation of Shanghai University of Medicine & Health Sciences (HMSF-16-21-007 and JW-2014ZZBJ-px(YX)-GY01) and the National Natural Science Foundation of China (81372177).
Author information
Authors and Affiliations
Contributions
Bin Hui, Liping Zhang, and Qinhua Zhou designed the experiments; Bin Hui, Qinhua Zhou, Liping Zhang, and Ling Hui performed parts of the experiments; Bin Hui, Qinhua Zhou, and Liping Zhang analyzed the data; Bin Hui, Qinhua Zhou, and Liping Zhang wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hui, B., Zhang, L., Zhou, Q. et al. Pristimerin Inhibits LPS-Triggered Neurotoxicity in BV-2 Microglia Cells Through Modulating IRAK1/TRAF6/TAK1-Mediated NF-κB and AP-1 Signaling Pathways In Vitro. Neurotox Res 33, 268–283 (2018). https://doi.org/10.1007/s12640-017-9837-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-017-9837-3