
Abstract
Inflammatory bowel disease (IBD) in humans is closely related to bacterial infection and the disruption of the intestinal barrier. Paeoniflorin (PF), a bioactive compound from Paeonia lactiflora Pallas plants, exerts a potential effect of anti-inflammatory reported in various researches. However, the effect of PF on intestinal barrier function and its related mechanisms has not been identified. Here, we investigate the PF potential anti-inflammatory effect on lipopolysaccharide (LPS)-stimulated human Caco-2 cell monolayers and explore its underlying key molecular mechanism. In this context, PF significantly increased TEER value, decreased intestinal epithelium FITC-dextran flux permeability, and restored the expressions of occludin, ZO-1, and claudin5 in LPS-induced Caco-2 cell. In vitro, treatment of PF significantly inhibited LPS-induced expression of cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), and matrix metalloproteinase-9 (MMP-9). In addition, we found that PF suppressed nuclear factor kappa B (NF-κB) signaling via activating the Nrf2/HO-1 signaling pathways in ILPS-stimulated Caco-2 cells. Our findings indicate that PF has an inhibitory effect on endothelial injury. Our findings suggested that PF has an anti-inflammatory effect in ILPS-stimulated Caco-2 cells, which might be a potential therapeutic agent against IBD and intestinal inflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The intestinal epithelium separates the intestinal mucosa from the lumen environment, which is consisted of a continuous proliferating and differentiating intestinal epithelial cells’ (IECs) monolayer [23, 24]. Damage of the IECs’ tight junction (TJ) barrier due to any pathogens contributes to the progress of inflammatory bowel disease (IBD) through increased intestinal permeability [7, 16]. Thus, it is necessary and particularly important to prevent gut diseases by maintaining IECs’ proper barrier function in animals.
TJ proteins are the junctional multiprotein complexes’ most important intercellular junctions of IECs, which seal the paracellular space among adjacent IECs [28]. TJ proteins regulate water, ion transport, and solutes through the paracellular pathway and prevention of immunogenic macromolecules [23, 28]. These multiprotein complexes only allow selective and controlled movements during the passive permeability process. The TJ proteins’ physiological functions are maintained by three crucial proteins including zonula occludens-1 (ZO-1), occludin, and claudins. Previous researches have certified that ZO-1 and occludin upregulation could inhibit the growth of intestinal permeability [1, 25, 33]. Thus, drugs that prevent TJ protein disruption of ZO-1 and occludin are widely considered an effective approach for intestinal disorder therapies.
Lipopolysaccharide (LPS) plays a crucial role in causing intestinal and systemic inflammatory reaction [9, 22]. Previous studies have demonstrated that LPS increases TJ permeability and disruption of the intestinal TJ barrier [10, 25, 26]. In addition, persistent inflammatory stimulation induces functional impairment of the intestinal TJ barrier to further generate pro-inflammatory cytokines which constitute a vicious cycle. Therefore, approaches that can alleviate inflammatory response may present promising strategies to be therapeutic interventions for IBD. Several in vitro tests have indicated that natural compounds such as vitamin A could reverse LPS-induced intestinal barrier damage [9], extracts of Boswellia serrata and Curcuma longa showed a good protective effect on the intestinal epithelium, with TEER values increasing compared with LPS-stimulated cells [6], the fixed combination of probiotics and herbal extracts prevented the inflammation-induced TEER decrease [5], and 6-gingerol could restore intestinal barrier function and suppress pro-inflammatory responses [2].
Paeoniflorin (PF, Fig. 1a) is the major bioactive compound isolated from Paeonia lactiflora Pallas, a traditional Chinese herb [30, 31]. It has been demonstrated to exhibit anti-inflammatory, antioxidative, and immunomodulatory effects [8, 12, 19]. However, to date, its anti-inflammatory effect specifically against intestinal inflammation has not been identified. The differentiated porcine intestinal epithelial cell line Caco-2 cell is a recognized cellular model stimulated with relevant cytokines or LPS to explore the role of the inflammation of intestinal barrier [3]. We used Caco-2 cells as a cellular model to investigate the protective effect of PF on intestinal barrier under the LPS-induced inflammation condition and to explore its underlying mechanism.

Dose-effect of PF on Caco-2 cells viability. a Chemical structure of PF. b, c The cytotoxic effects of PF on the Caco-2 cells’ viability were assessed at increasing concentrations of PF (0, 10, 50, 100, and 150 μM) for 24 or 48 h by CCK8 assay. The values presented are the means ± S.D. *P < 0.05 vs. control group, n = 5.
MATERIALS AND METHODS
Materials
Paeoniflorin was purchased from Solarbio (Beijing, China). LPS was derived from Salmonella enterica ser. and obtained from Sigma-Aldrich (St Louis, MO, USA). Cell culture mediums containing fetal bovine serum (FBS), trypsin/EDTA, and antibiotics (penicillin/streptomycin solution) were supplied by GIBCO (Carlsbad, CA, USA). Antibodies against occludin, Claudin5, and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-HO-1, COX-2, MMP-9, lamin B, and ZO-1 were supplied by Cell Signaling Technologies (Danvers, MA, USA). Anti-iNOS and Nrf2 were purchased from Abcam. Anti-NF-κB and iκBα were obtained from Bioworld (OH, USA). The secondary antibodies conjugated with goat anti-rabbit IgG and Alexa Fluor® 488 were obtained from Abcam. All other chemicals were from Sigma.
Cell Culture
Caco-2 cells were grown in 37 °C with 5% CO2 in DMEM/F12 medium containing 10% FBS and 1% penicillin-streptomycin antibiotics. The medium was changed every 2 days. Cells were starved in serum-free medium before each experiment for 24 h.
Cell Viability Assay
The PF cytotoxicity to chondrocytes was evaluated using a CCK-8 assay (Dojindo Co., Japan) according to the manufacturer’s instructions. Caco-2 cells were seeded in 96-well plates at a density of 2 × 104/well and cultured for 24 h. Then, cells were treated with LPS (10 μg mL−1) or PF at different concentrations of 10, 50, 100, and 150 μM for 24 h and 48 h. Then, 10 μL CCK-8 solution was added to each plate and further cultured for 2 h; after that, the absorbance was measured at 450-nm wavelength using a micro-plate reader. Each experiment was carried out five times.
Measurement of TEER and Permeability
Transepithelial resistance (TEER) was measured to assess the barrier integrity of Caco-2 cells. Caco-2 cells were seeded in a transwell chamber with 4.5-μm pores (Costar, NY, USA) that had been placed in a 6-well plate at a density of 6 × 105 cells/well. Cells were cultured for 7–9 days in the medium and completely differentiated. TEER was performed to measure cell monolayer integrity before and after all treatments. The TEER values were measured using an ohmmeter with chopstick electrodes (Millipore ESR-2; Burlington, MA, USA). The data were presented as unit area resistance calculated by dividing resistance values by the effective membrane area. Inserts without cells were used as blanks.
Permeability of the monolayer Caco-2 cell measurements were performed by the flux of FITC-dextran (4 kDa, Sigma-Aldrich). FITC-dextran (1 mg/mL) was added to the apical compartment of the insets. After 6 h of incubation, the basolateral medium aliquots were collected for the measurement of fluorescence at 480-nm excitation and 520-nm emission wavelengths.
RT-PCR
Total RNA was isolated using TRIzol reagent on the basis of the manufacturer’s protocol. Briefly, cDNA was synthesized from 1000 ng of total RNA (MBI Fermantas, Germany). The parameters of RT-PCR were 10 min 95 °C, then 95 °C for 15 s, and 1 min at 60 °C, performed for 40 cycles. The reaction was performed via a CFX96 Real-Time PCR System (Bio-Rad Laboratories, CA, USA). The cycle threshold (Ct) values were calculated and normalized to the level of a housekeeping gene GAPDH. The forward and reverse primers are shown as follows: TNF-α (F) 5′-GTCAGATCATCTTCTCGA ACC-3′, (R) 5′-CAGATAGATGGGCTCATACC-3′; IL-6 (F) 5′-GACAGCCACTCACCTCTTCA-3′, (R) 5′-TTCACCAGGCAAGTCTCCTC-3′; COX-2 (F) 5′-GAGAGATGTATCCTCCCACAGTCA-3′ (R) 5′-GACCAGGCACCAGACCAAAG-3′; iNOS (F) 5′-CCTTACGAGGCGAAGAAGGACAG-3′, (R) 5′-CAGTTTGAGAGAGGAGGCTCCG-3′; occludin (F) 5′-GAGTTGTATCTGTTGTTGT-3′ (R) 5′-TTCGTGGTATAGCATTCT-3′; ZO-1 (F) 5′-GGTGAAGTGAAGACAATG-3′, (R) 5′-GGTAATATGGTGAAGTTAGAG-3′; Claudin-5 (F) 5′-TTAACAGACGGAATGAAGT-3′ (R) 5′-GAAGCGAAATCCTCAGTC-3′;
Western Blotting
Total proteins were extracted from Caco-2 cells with a lysis buffer. Then, the protein concentrations were tested by using a BCA protein assay kit (Beyotime). Forty micrograms of protein sample was separated by SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, USA). After blocking with 5% nonfat milk for 1.5 h, the membrane was incubated with solutions of the appropriate primary antibodies overnight at 4 °C. Then, incubation with the respective horseradish peroxidase (HRP)–conjugated secondary antibody at room temperature for 2 h. After washing with TBST 3 times, the blots were visualized via electrochemiluminescence plus reagent (Invitrogen). Band intensity was quantified using Image Lab software (Bio-Rad). The experiment was repeated five times.
Assay of Cell Immunofluorescence
Cells were cultured onto cover slips in a 6-well plate for 24 h. Then, cells were washed with PBS 3 times and fixed with 4% paraformaldehyde for 15 min and blocked with 5% bovine serum albumin in a wet box at 37 °C for 1 h. After cells were rinsed with PBS, the primary antibodies such as occludin (1: 300) and p65 (1: 300) were added and incubated with the cells in a humidity chamber at 4 °C overnight. The slides were then rinsed with PBS 3 times, incubated with Alexa Fluor®594-labeled or Alexa Fluor®488-labeled secondary antibodies (1: 500) for 2 h at room temperature. Then, cell nuclei were stained by DAPI. Finally, the slides were observed with a fluorescence microscope. Five visual fields of each slides were randomly selected microscopic observation.
siRNA Transfection
Nrf2 small-interfering RNA (siRNA) was obtained from Invitrogen (Carlsbad, CA, USA). The Nrf2 siRNA sequences were as follows: sense strand 5′-GUAAGAAGCCAGAUGUUAAdUdU-3′. Lipofectamine™ RNAiMAX reagent was used to transfect Nrf2 and negative control (NC) siRNA on the basis of the manufacturer’s protocol.
Statistical Analysis
Data are expressed as the mean ± SEM. Statistics was analyzed via GraphPad Prism version5 (GraphPad Software, San Diego, CA). One-way ANOVA followed by Tukey’s test was used to compare differences among each group. Level of significance was set at p < 0.05.
RESULTS
Dose-Effect of PF on Caco-2 Cell Viability
The chemical structure of PF is shown in Fig. 1a. The cytotoxic effects of PF on the Caco-2 cell viability were assessed at increasing concentrations of PF (0, 10, 50, 100, and 150 μM) for 24 or 48 h. Then, CCK-8 assay was used to detect the cellular viability.
The results of CCK8 imply that PF decreased cell viability significantly at 150 μM compared with untreated cells (P < 0.05) after 48 h of culturing. It suggested that PF had no cytotoxicity to Caco-2 cells after 24 h or 48 h at concentrations ≤ 150 μM. Therefore, we used PF with the doses of 10, 50, and 100 μM in our subsequent experiments.
PF Inhibits LPS-Induced Inflammation Reaction in Caco-2 Cells
To explore the PF anti-inflammatory effect on Caco-2 cells, alterations of IL-6, iNOS, TNF-α, and COX-2 were examined by RT-PCR or Western blotting. As is shown in Fig. 2a–d, LPS increased the mRNA levels expression of IL-6, iNOS, TNF-α, and COX-2, whereas PF reversed these upregulations in a dose-dependent pattern. We next detected the effect of PF on COX-2 and iNOS production in protein level by Western blotting. Results showed that LPS upregulated the production of iNOS and COX-2 at protein levels and PF inhibited these increases in a dose-dependent manner (50 and 100 μM) (Fig. 2e–g), but no statistical significance was found in the 10-μM treatment group. The above data suggest that PF prominently inhibited the production of LPS-induced inflammatory cytokines and mediators at the levels of gene and protein in a concentration-dependent pattern.

PF inhibits LPS-induced inflammation reaction in Caco-2 cells. a–d The mRNA expressions of iNOS, IL-6, COX-2, and TNF-α were detected using real-time PCR. e iNOS and COX-2 protein expressions in Caco-2 cells were determined via Western blotting and f, g quantified. The data in the figures represent mean ± S.D. ##P < 0.01 compared with the control group; *P < 0.05, **P < 0.01 compared with the LPS alone group, n = 5.
PF Prevents the Loss of TJ Protein in LPS-Induced Caco-2 Cells
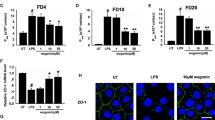
TEER was used to measure the monolayer integrity of Caco-2 cells. TEER values were assessed at 12, 24, 36, and 48 h after LPS or PF (100 μM) administration. As is shown in Fig. 3a, LPS induced the decrease of TEER dramatically after 24-h treatments, continuing to 48 h after administration. On the contrary, PF significantly increased TEER value, suggesting that PF decreased intestinal epithelium permeability. Moreover, we detected large solutes permeability of Caco-2 cells by using FITC-dextran flux experiment. The result showed that the FITC-dextran concentrations in the basal compartment were dramatically higher after cell incubation with FITC-dextran in the LPS group than that the in control group. PF treatment significantly reduced the leakage of FITC-dextran induced by LPS stimulation compared with the LPS application group (Fig. 3b). LPS respectively decreased proteins (Fig. 3c–f). The treatment of PF restored the expressions of occludin, ZO-1, and claudin5 (P < 0.05 vs. LPS). In addition, the result of occludin immunofluorescence analysis was in accordance with the Western blot results (Fig. 3g). Altogether, these data suggested that PF preserved Caco-2 cell integrity by inhibiting the loss of TJ proteins in LPS-induced Caco-2 cells.

PF at 100-μM concentration prevents the loss of TJ protein in LPS-induced Caco-2 cells. a TEER values were assessed at 12, 24, 36, and 48 h after LPS or PF administration. b Caco-2 cell large solute permeability was detected in each group by using FITC-dextran flux experiment. c–f The protein expressions of occludin, ZO-1, and claudin5 in Caco-2 cells treated as above were detected by Western blot. The representative occludin was visualized by the immunofluorescence analysis and DAPI staining of nuclei in Caco-2 cells (scale bar 20 μm). The data in the figures represent mean ± S.D. ##P < 0.01 compared with the control group; *P < 0.05, **P < 0.01 compared with the LPS administration group, n = 5.
PF Regulates the Intestinal Barrier by Inhibiting LPS-Induced NF-κB Activation
We next investigated the NF-κB (p65) pathway in Caco-2 cells by using Western blot analysis. The p65 expression in the nucleus increased and the IκBα expression in the cytoplasm decreased after LPS stimulation, whereas the pretreatment of PF (100 μM) for 24 h inhibited significantly the above response (Fig. 4a–d). PF pretreatment also significantly inhibited MMP9 expression in LPS-induced Caco-2 cells (Fig. 4a, b). These phenomena were because of the translocation of p65 from the cytoplasm to the nuclei. Immunofluorescence staining of p65 was performed to evaluate its translocation. The results showed that in the control group, p65 mostly localized in the cytoplasm. p65 staining was intense in the nucleus following LPS stimulation. PF pretreatment for 24 h dramatically inhibited the LPS-induced p65 nuclear translocation, which was in accordance with the Western blotting result. These results indicated that PF dramatically inhibited LPS-induced NF-κB activation in Caco-2 cells.

PF (100 μM) regulates the intestinal barrier by inhibiting LPS-induced NF-κB activation. The protein expressions of MMP-9, p65 in nuclear, and IκBα in cytoplasm in Caco-2 cells in each group were a measured by Western blotting, and b–d quantified. Representative micrographs showing immunofluorescence staining with p65 (green) nuclei are (e) labeled with DAPI in each group. Values represent the mean ± S.D. ##P < 0.01 vs. control group; *P < 0.05, **P < 0.01 as compared with the cells treated with LPS, n = 5.
The Effect of PF on the Nrf2/HO-1 Signal Pathway
Nrf2 activation inhibits the inflammatory reaction. We next assessed Nrf2 and HO-1 protein levels via Western blotting. The expression of abundance of Nrf2 and HO-1 proteins were illustrated in Fig. 5a–c. Results showed that Nrf2 and HO-1 protein expression increased following the PF (100 μM) treatment under LPS stimulation. These data suggested that PF enhanced Nrf2 protein expression. RNA interference (RNAi) against Nrf2 was performed to further clarify Nrf2/HO-1 signal pathway mediates PF-induced NF-κB signal inhibition. Western blot results suggested that the expressions of nuclear Nrf2 and cytoplasmic HO-1 were significantly inhibited in LPS and PF (100 μM) co-treated Caco-2 cells after Nrf2 siRNA transfection (Fig. 5d–f). Furthermore, we examined the TEER values and diffusion of FITC-dextran after Nrf2 siRNA transfection. As is shown in Fig. 5 g and h, Nrf2 siRNA abolished the PF-induced increase of TEER values and the decrease of after the FITC-dextran diffusion in LPS administration group cells.

The effect of PF on the Nrf2/HO-1 signal pathway. a–c Representative Western blots and quantification data of Nrf2 in the nucleus and HO-1 in the cytoplasm in Caco-2 cells. d, f After Nrf2 knockdown, HO-1 in the cytoplasm and Nrf2 in the nucleus were assessed by Western blotting analysis, **P < 0.01. g TEER values in each group were assessed at 12, 24, 36, and 48 h. h Caco-2 cells large solute permeability was detected in each group by using FITC-dextran flux experiment. Values represent the mean ± S.D. ##P < 0.01 vs. LPS group, *P < 0.05, **P < 0.01 as compared with PF at the 100-μM concentration–treated group, n = 5.
Involvement of the Nrf2/HO-1 Pathway in the Effect of PF on Regulation of Intestinal Barrier and NF-κB Activation
As is shown in Fig. 6a–c, the TJ proteins’ protective effect of PF (100 μM) was significantly inhibited after the transfection of Nrf2 siRNA under inflammatory conditions. The mRNA expressions of ZO-1, occludin, and claudin5 were significantly decreased in the Nrf2 siRNA transfection group compared with the PF treatment group. On the contrary, expression of nuclear p65 was increased significantly in the Nrf2 siRNA transfection group, which suggested that the Nrf2 pathway mediates the inhibition of NF-κB activation effect of PF on Caco-2 cells under inflammatory conditions (Fig. 6d, e). In addition, we detected the mRNA expressions of IL-6, iNOS, TNF-α, and COX-2 and found that Nrf2 siRNA transfection abolished the PF-mediated effect of anti-inflammatory (Fig. 6f–i). Altogether, these results suggest that the Nrf2 signaling pathway plays a protective role in PD treatment in IL-1β-stimulated human OA chondrocytes.

Involvement of the Nrf2/HO-1 pathway in the effect of PF at 100-μM concentration on regulation of intestinal barrier and NF-κB activation. a–c The mRNA expressions of occludin, ZO-1, and claudin5 in Caco-2 cells treated as above were detected by RT-PCR after Nrf2 knockdown. d, e p65 in nucleus was assessed by Western blotting analysis following Nrf2 knockdown. f–i The mRNA expressions of iNOS, IL-6, COX-2, and TNF-α were detected using real-time PCR in Caco-2 cells treated as above after Nrf2 knockdown. Values represent the averages ± S.D. **P < 0.01, n = 5.
DISCUSSION
IBD, including ulcerative colitis and Crohn’s disease, is gastrointestinal tract chronic inflammatory disorder characterized by remission and periods, which influence millions of personal and family life quality in developed countries all over the world [18]. IBD etiology remains not yet completely understood, and current treatment is far from satisfactory, not only showing high ineffectiveness but also showing serious side effects in a mass of patients [20, 21]. Therefore, new intervention and therapeutic strategies are needed to handle these inflammatory disorders.
The previous study has revealed that PF can remarkably attenuate inflammatory response in vascular disease [4]. However, the effect of PF on inflammatory response in IECs under damage condition remains unknown. Our results of CCK8 implied that PF decreased cell viability significantly at 150 μM compared with untreated cells (P < 0.05) after 48 h of culturing. It suggested that PF had no cytotoxicity to Caco-2 cells after 24 h or 48 h at concentrations ≤ 150 μM. Results also showed that PF inhibited iNOS, and COX-2 increases in a dose-dependent manner (50 and 100 μM) in LPS stimulation. Therefore, PF with the doses of 100 μM in our subsequent experiments was used. The major finding of our study is that PF attenuates LPS-induced Caco-2 cell inflammatory reaction and TJ protein disruption by inhibiting the NF-κB pathway and activating the pathway of Nrf2/HO-1. Most importantly, the NF-κB inhibition effect of PF was likely achieved by the Nrf2 activation. The above results suggest that the use of PF can be a potential anti-inflammatory agent for the prevention of the context of IBD.
It is widely believed that the destruction of the intestinal epithelial TJ barrier leads to increased intestinal permeability and plays a key role in the progress of IBD. TJ proteins play a critical role in maintaining the intestinal epithelial barrier function under oxidative stress or inflammatory conditions because they mechanically seal the transparency between IECs [1, 25, 33]. Notably, experimental data of colon biopsies of IBD patients highlighted an impairment of the intestinal barrier accompanied by a decreased expression of TJ proteins [11, 32]. These facts suggest that regulating TJ proteins can promote the integrity of the intestinal barrier and may constitute an effective therapeutic strategy for IBD. In the present study, TEER and cell monolayer permeability experiments were performed to assess the intestinal barrier function [5]. The results showed that PF could remarkably increase TEER value and reduce FITC-dextran flux permeability in LPS-induced Caco-2 cell inflammatory conditions. We also found that LPS diminished the key sealing TJ protein expression, such as ZO-1, occluding, and claudin-5 and PF inflammation upregulated the level of TJ mRNA and proteins. Our data suggest that PF may regulate the barrier function by regulating TJ protein expression.
Numerous evidences have revealed that the NF-κB pathway plays a critical role in the pathogenesis and progression of IBD [20, 21]. LPS stimulation triggers IκBα phosphorylation leading to IκBα degradation, which consequently results in free and translocation of NF-κB from cytoplasm to nucleus. In the nucleus, p65 upregulates the transcription of pro-inflammatory mediators and cytokines, for instance, COX-2, TNF-α, iNOS, and IL-6. iNOS could catalyze the guanidine nitrogen of l-arginine leading to the production of NO, which stimulates the secretion of MMPs resulting in TJ protein disruption. Previous researches have demonstrated that PF inhibited vascular inflammatory reaction by suppressing the TLR-4/NF-κB signaling pathway and partly blocking the LPS-stimulated endothelial permeability [29, 30, 34]. The NF-κB signaling pathway was detected in the present study to illuminate PF mechanism in regulating the intestinal monolayer barrier. The results showed that LPS-induced NF-κB activation in Caco-2 cells was inhibited by PF. Moreover, PF reversed the upregulations of LPS-increased IL-6, iNOS, TNF-α, and COX-2 expression in a dose-dependent pattern at the mRNA and protein level.
Several researchers have suggested the activation of Nrf2 contributes to suppressing the inflammatory process in vivo and in vitro models, and the inhibition of Nrf2 increases the susceptibility to inflammatory disease [15, 17]. HO-1 is a downstream target protein of Nrf2 and degrades heme to liberate biliverdin, CO, and ferrous ion (Fe2+). Previous studies have indicated that the Nrf2/HO-1 pathway has assumed one of the upstream molecules considered targets for anti-NF-κB-induced inflammation [13, 14, 27]. In the current study, we found that inflammatory response was obviously inhibited by PF treatment-induced Nrf2 activation while the TJ proteins were upregulated under LPS-induced inflammatory conditions. However, the Nrf2-siRNA strongly prevented the PF-mediated inhibition of the LPS-induced translocation of NF-κB and expression of inflammatory mediators in Caco-2 cells, supporting that PF inhibited the NF-κB-dependent inflammatory response by activating the Nrf2/HO-1 pathway. Taken together, the above data reveal that Nrf2/HO-1 is involved in the PF protective effect on Caco-2 cells.
In conclusion, the current research has exhibited that PF dramatically decreased the inflammatory response and TJ protein destruction induced by LPS by inhibiting NF-κB activation and stimulating the Nrf2-dependent HO-1 pathway (Fig. 7) in mouse Caco-2 cells. Overall, these data may contribute to ascertain the use of PF as a therapeutic strategy for the inflammation-associated disorder management in IBD patients.

A model illustration potential molecular mechanism involved in the PF-treated Caco-2 cells.
References
Bazzoni, Gianfranco, and Elisabetta Dejana. 2004. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiological Reviews 84 (3): 869–901.
Chang, K.W., and C.Y. Kuo. 2015. 6-Gingerol modulates proinflammatory responses in dextran sodium sulfate (DSS)-treated Caco-2 cells and experimental colitis in mice through adenosine monophosphate-activated protein kinase (AMPK) activation. Food & Function 6 (10): 3334–3341. https://doi.org/10.1039/c5fo00513b.
Chen, Qianru, Oliver Chen, Isabela M. Martins, Hou Hu, Zhao Xue, Jeffrey B. Blumberg, and Bafang Li. 2017. Collagen peptides ameliorate intestinal epithelial barrier dysfunction in immunostimulatory Caco-2 cell monolayers via enhancing tight junctions. Food & Function 8 (3): 1144–1151. https://doi.org/10.1039/c6fo01347c.
Chen, J., M. Zhang, M. Zhu, J. Gu, J. Song, L. Cui, D. Liu, Q. Ning, X. Jia, and L. Feng. 2018. Paeoniflorin prevents endoplasmic reticulum stress-associated inflammation in lipopolysaccharide-stimulated human umbilical vein endothelial cells via the IRE1alpha/NF-kappaB signaling pathway. Food & Function 9 (4): 2386–2397. https://doi.org/10.1039/c7fo01406f.
Cocetta, V., D. Catanzaro, V. Borgonetti, E. Ragazzi, M.C. Giron, P. Governa, I. Carnevali, M. Biagi, and M. Montopoli. 2019. A fixed combination of probiotics and herbal extracts attenuates intestinal barrier dysfunction from inflammatory stress in an in vitro model using Caco-2 cells. Recent Patents on Food, Nutrition & Agriculture 10 (1): 62–69. https://doi.org/10.2174/2212798410666180808121328.
Governa, P., M. Marchi, V. Cocetta, B. De Leo, P.T.K. Saunders, D. Catanzaro, E. Miraldi, M. Montopoli, and M. Biagi. 2018. Effects of Boswellia Serrata Roxb. and Curcuma longa L. in an in vitro intestinal inflammation model using immune cells and Caco-2. Pharmaceuticals (Basel) 11 (4). https://doi.org/10.3390/ph11040126.
Guan, Qingdong, and Jiguo Zhang. 2017. Recent advances: The imbalance of cytokines in the pathogenesis of inflammatory bowel disease. Mediators of Inflammation 2017: 1–8.
Guo, Ruo-Bing, Guo-Feng Wang, An-Peng Zhao, Gu Jun, Xiu-Lan Sun, and Hu. Gang. 2012. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-κB-mediated inflammatory responses. PLoS One 7 (11): e49701.
He, C., J. Deng, X. Hu, S. Zhou, J. Wu, D. Xiao, K.O. Darko, Y. Huang, T. Tao, M. Peng, Z. Wang, and X. Yang. 2019. Vitamin A inhibits the action of LPS on the intestinal epithelial barrier function and tight junction proteins. Food & Function 10 (2): 1235–1242. https://doi.org/10.1039/c8fo01123k.
He, Caimei, Jun Deng, Xin Hu, Sichun Zhou, Jingtao Wu, Di Xiao, Kwame Oteng Darko, Yanjun Huang, Ting Tao, and Mei Peng. 2019. Vitamin A inhibits the action of LPS on the intestinal epithelial barrier function and tight junction proteins. Food & Function 10 (2): 1235–1242.
Heller, Frank, Peter Florian, Christian Bojarski, Jan Richter, Melanie Christ, Bernd Hillenbrand, Joachim Mankertz, Alfred H. Gitter, Nataly Bürgel, and Michael Fromm. 2005. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129 (2): 550–564.
Jiang, Zequn, Weiping Chen, Xiaojing Yan, Lei Bi, Sheng Guo, and Zhen Zhan. 2014. Paeoniflorin protects cells from GalN/TNF-α-induced apoptosis via ER stress and mitochondria-dependent pathways in human L02 hepatocytes. Acta Biochimica et Biophysica Sinica 46 (5): 357–367.
Kim, Y.J., and W. Park. 2016. Anti-inflammatory effect of quercetin on RAW 264.7 mouse macrophages induced with polyinosinic-polycytidylic acid. Molecules 21 (4): 450. https://doi.org/10.3390/molecules21040450.
Lee, J.M., J. Li, D.A. Johnson, T.D. Stein, A.D. Kraft, M.J. Calkins, R.J. Jakel, and J.A. Johnson. 2005. Nrf2, a multi-organ protector? The FASEB Journal 19 (9): 1061–1066. https://doi.org/10.1096/fj.04-2591hyp.
Lee, D.F., H.P. Kuo, M. Liu, C.K. Chou, W. Xia, Y. Du, J. Shen, et al. 2009. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Molecular Cell 36 (1): 131–140. https://doi.org/10.1016/j.molcel.2009.07.025.
Lee, Seung Hoon, Jeong eun Kwon, and Mi-La Cho. 2018. Immunological pathogenesis of inflammatory bowel disease. Intestinal Research 16 (1): 26–42.
Liu, G.H., J. Qu, and X. Shen. 2008. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochimica et Biophysica Acta 1783 (5): 713–727. https://doi.org/10.1016/j.bbamcr.2008.01.002.
Malik, Talha A. 2015. Inflammatory bowel disease: Historical perspective, epidemiology, and risk factors. Surgical Clinics 95 (6): 1105–1122.
Nam, Kyong-Nyon, Che Gyem Yae, Joung-Woo Hong, Dong-Hyung Cho, Joon H. Lee, and Eunjoo H. Lee. 2013. Paeoniflorin, a monoterpene glycoside, attenuates lipopolysaccharide-induced neuronal injury and brain microglial inflammatory response. Biotechnology Letters 35 (8): 1183–1189.
Nunes, Carla, Leonor Almeida, Rui M. Barbosa, and João Laranjinha. 2017. Luteolin suppresses the JAK/STAT pathway in a cellular model of intestinal inflammation. Food & Function 8 (1): 387–396. https://doi.org/10.1039/c6fo01529h.
Nunes, C., V. Freitas, L. Almeida, and J. Laranjinha. 2019. Red wine extract preserves tight junctions in intestinal epithelial cells under inflammatory conditions: Implications for intestinal inflammation. Food & Function 10 (3): 1364–1374. https://doi.org/10.1039/c8fo02469c.
Omonijo, F.A., S. Liu, Q. Hui, H. Zhang, L. Lahaye, J.C. Bodin, J. Gong, M. Nyachoti, and C. Yang. 2019. Thymol improves barrier function and attenuates inflammatory responses in porcine intestinal epithelial cells during lipopolysaccharide (LPS)-induced inflammation. Journal of Agricultural and Food Chemistry 67 (2): 615–624. https://doi.org/10.1021/acs.jafc.8b05480.
Pitman, Richard S., and Richard S. Blumberg. 2000. First line of defense: The role of the intestinal epithelium as an active component of the mucosal immune system. Journal of Gastroenterology 35 (11): 805–814.
Sartor, R. Balfour. 2006. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nature Reviews Gastroenterology & Hepatology 3 (7): 390.
Siliciano, J.D., and Daniel A. Goodenough. 1988. Localization of the tight junction protein, ZO-1, is modulated by extracellular calcium and cell-cell contact in Madin-Darby canine kidney epithelial cells. The Journal of Cell Biology 107 (6): 2389–2399.
Tang, X., B. Liu, X. Wang, Q. Yu, and R. Fang. 2018. Epidermal growth factor, through alleviating oxidative stress, protect IPEC-J2 cells from lipopolysaccharides-induced apoptosis. International Journal of Molecular Sciences 19 (3). https://doi.org/10.3390/ijms19030848.
Tenhunen, R., H.S. Marver, and R. Schmid. 1968. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proceedings of the National Academy of Sciences of the United States of America 61 (2): 748–755.
Tsukita, Shoichiro, Mikio Furuse, and Masahiko Itoh. 2001. Multifunctional strands in tight junctions. Nature Reviews Molecular Cell Biology 2 (4): 285–293.
Wu, Y.-M., R. Jin, L. Yang, J. Zhang, Q. Yang, Y.-Y. Guo, X.-B. Li, S.-B. Liu, X.-X. Luo, and M.-G. Zhao. 2013. Phosphatidylinositol 3 kinase/protein kinase B is responsible for the protection of paeoniflorin upon H2O2-induced neural progenitor cell injury. Neuroscience 240: 54–62.
Xu, Huan, Jie Song, Xinghua Gao, Zhao Xu, Xianxiang Xu, Yufeng Xia, and Yue Dai. 2013. Paeoniflorin attenuates lipopolysaccharide-induced permeability of endothelial cells: Involvements of F-actin expression and phosphorylations of PI3K/Akt and PKC. Inflammation 36 (1): 216–225.
Yin, Dou, Yuan-Yuan Liu, Tian-Xiao Wang, Zhen-Zhen Hu, Qu Wei-Min, Jiang-Fan Chen, Neng-Neng Cheng, and Zhi-Li Huang. 2016. Paeoniflorin exerts analgesic and hypnotic effects via adenosine A 1 receptors in a mouse neuropathic pain model. Psychopharmacology 233 (2): 281–293.
Zeissig, Sebastian, Nataly Bürgel, Dorothee Günzel, Jan Richter, Joachim Mankertz, Ulrich Wahnschaffe, Anton Josef Kroesen, Martin Zeitz, Michael Fromm, and Joerg Dieter Schulzke. 2007. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56 (1): 61–72.
Zhang, Bingkun, and Yuming Guo. 2009. Supplemental zinc reduced intestinal permeability by enhancing occludin and zonula occludens protein-1 (ZO-1) expression in weaning piglets. British Journal of Nutrition 102 (5): 687–693.
Zhou, J., L. Wang, J. Wang, C. Wang, Z. Yang, C. Wang, Y. Zhu, and J. Zhang. 2016. Paeoniflorin and albiflorin attenuate neuropathic pain via MAPK pathway in chronic constriction injury rats. Evidence-based Complementary and Alternative Medicine 2016: 8082753–8082711. https://doi.org/10.1155/2016/8082753.
Funding
This work was financially supported by Grants from National Natural Science Foundation of China (81570495).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, XX., Huang, XL., Chen, RR. et al. Paeoniflorin Prevents Intestinal Barrier Disruption and Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in Caco-2 Cell Monolayers. Inflammation 42, 2215–2225 (2019). https://doi.org/10.1007/s10753-019-01085-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01085-z