
Abstract
Diabetes is a global epidemic and a leading cause of death with more than 422 million patients worldwide out of whom around 392 million alone suffer from type 2 diabetes (T2D). Sodium-glucose cotransporter 2 inhibitors (SGLT2i) are novel and effective drugs in managing glycemia of T2D patients. These inhibitors gained recent clinical and basic research attention due to their clinically observed cardiovascular protective effects. Although interest in the study of various SGLT isoforms and the effect of their inhibition on cardiovascular function extends over the past 20 years, an explanation of the effects observed clinically based on available experimental data is not forthcoming. The remarkable reduction in cardiovascular (CV) mortality (38%), major CV events (14%), hospitalization for heart failure (35%), and death from any cause (32%) observed over a period of 2.6 years in patients with T2D and high CV risk in the EMPA-REG OUTCOME trial involving the SGLT2 inhibitor empagliflozin (Empa) have raised the possibility that potential novel, more specific mechanisms of SGLT2 inhibition synergize with the known modest systemic improvements, such as glycemic, body weight, diuresis, and blood pressure control. Multiple studies investigated the direct impact of SGLT2i on the cardiovascular system with limited findings and the pathophysiological role of SGLTs in the heart. The direct impact of SGLT2i on cardiac homeostasis remains controversial, especially that SGLT1 isoform is the only form expressed in the capillaries and myocardium of human and rodent hearts. The direct impact of SGLT2i on the cardiovascular system along with potential lines of future research is summarized in this review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
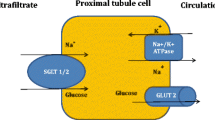
Diabetes is a chronic disease characterized by the inability of the body to either produce enough insulin or effectively employ the insulin it produces to control blood glucose levels [1]. Uncontrollable and sustained increase in blood glucose levels could seriously damage vital organs and systems such as the heart, kidneys, blood vessels, eyes, and nerves. Multiple complications such as coronary artery disease, stroke, nephropathy, neuropathy, and retinopathy are directly linked to long-term diabetes and negatively impact the lifespan of the diabetic population [2]. In 2012, diabetes was the 8th cause of death worldwide for both sexes with a total estimate of 3.7 million deaths, out of which 1.5 million were directly related to diabetes while the other 2.2 million deaths were linked to high blood glucose levels. According to the world health organization (WHO), the number of worldwide patients with diabetes increased between 1980 and 2014 from 108 million to 422 million with a prevalence of 8.5% among the adult population (WHO, Global report on diabetes, 2016). Diabetes is projected to become the 7th leading cause of death by 2030 [3]. Although long-known anti-diabetic drugs such as metformin, sulfonylureas, meglitinides, thiazolidinediones, and dipeptidyl-peptidase-4 inhibitors have been effective in lowering glucose levels independently or in combination therapy, they show no reduction in adverse cardiovascular outcomes and are associated with multiple side effects including hypoglycemia, weight gain, fluid retention, and increased risk of congestive heart failure [4,5,6,7]. SGLT2 inhibitors (SGLT2i) recently emerged as promising antidiabetic drugs with high therapeutic index and effectively lower blood glucose levels in type II diabetes (T2D) populations [8]. SGLT is a sodium-glucose cotransporter detected in two major isoforms, SGLT1 and SGLT2. The latter is mainly expressed in the lumen of the small intestine and kidneys and is involved in the absorption/reabsorption of glucose driven by the sodium gradient across the cell membrane [8, 9]. SGLT1 expression on the other hand was mainly detected in other tissues of the cardiovascular system (CV) including cardiac capillaries and cardiomyocytes of human and rodent heart, playing an important role in glucose uptake into the myocardium [9,10,11,12]. Emerging clinical evidence reports reduced negative CV outcomes with SGLT2 inhibitor therapy [13]. Yet, an equally interesting observation is the positive effect on BP reduction, arterial stiffness, vascular resistance, and microvascular remodeling [14,15,16,17]. Whether SGLT2 inhibition in T2D patients exerts any short- or long-term CV complications remain controversial. The clinical utility and the long-term CV outcomes of SGLT2i use in diabetic patients are subject to extensive research. Although experimental studies did not document SGLT2 expression in the heart, the positive impacts of SGLT2i on the heart are intriguing. The present review aims at exploring the potential mechanisms through which these drugs directly affect the CV system independently of their well-known systemic effects.
Diabetes and the emergence of SGLT2 inhibitors cardiovascular protection
Type 1 and type 2 diabetes are the most common types of diabetes. Type 1 diabetes (T1D), previously known as juvenile diabetes, accounts for 5 to 10% of diabetic cases and is characterized by abnormalities in sufficient insulin production with undefined mechanisms, mostly linked to both genetic and environmental factors [18]. In order to survive, T1D patients require a daily administration of insulin. T2D on the other hand is the most prevalent form of diabetes and constitutes 85 to 90% of all diabetic cases and results from the inability of the body to effectively use insulin [1, 19]. Unlike T1D, symptoms arising with T2D are less marked and mostly latent making diagnosis difficult and at later stages of the disease when serious complications emerge, unless high risk factors such as obesity are present [1].
Management of blood glucose levels in T2D patients presents a frequent and progressive challenge. As the most recent class of oral anti-hyperglycemic medications, SGLT2i provide a solution for a number of unmet clinical needs. Body weight, blood pressure (BP), and lipid reduction in addition to durable glycemic control were all much welcomed effects of the drug class in this patient cohort [20, 21]. Affecting metabolism independently of insulin without increasing hypoglycemic risk encouraged the use of SGLT2i in double or triple combination therapy with standard agents such as metformin and dipeptidyl peptidase-4 inhibitor, throughout the development of T2D [22].
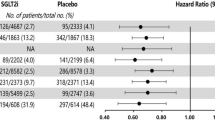
Typically, the detrimental vascular phenotype with T2D was thought to be a consequence of hyperglycemia through multiple and complex pathways [23]. Thus, current practice for management of patients with T2D focuses on maintaining good control of blood glucose level with a glycosylated hemoglobin target (HbA1c) < 7% [24]. This recommendation stems from robust clinical evidence documenting a reduced rate of development of micro-vascular complications (i.e., diabetic neuropathy, retinopathy, nephropathy) with good glycemic control in T2D patients, which initiated during the interventional trial and was sustained for years throughout the post-trial period [25, 26]. Follow-up clinical studies showed that intensive glucose control at the time of T2D diagnosis not only protects against microvascular complications in early and post-trial stages, but also extends to protection from macrovascular complications (i.e., CAD, PAD), which emerge overtime and in the post-trial follow-up phase only [26]. This led to the emergence of the “Legacy Effect” or “Cardiovascular Metabolic Memory” concept whereby the extent of vascular damage is determined, not only by current glycemic control, but also by the history of blood glucose level management [27]. To date, no promising drug that target diabetes has proven effective in preventing adverse cardiac remodeling following cardiac injury such as myocardial infarction (MI), independently of glycemic control [28, 29]. However, emerging evidence supports a protective effect for certain anti-hyperglycemic drugs against CV complications in diabetic patients already receiving standard of care for glycemic control and cardiovascular disease (CVD) [30, 31]. An exciting aspect of SGLT2i therapeutic effect in T2D patients is potential direct cardioprotection independently of glycemic control. SGLT2i, empagliflozin (Empa), is the first drug to display significant cardioprotection in clinical trials with a clear reduction in CV mortality and hospitalization due to heart failure within the first 3 months of initiating the treatment [31]. Speculations on the basis of such effects included a role for the observed reduction of BP, body weight, and increased diuretic effects (see box for EMPA-REG Outcome Trial Summary) [32]. Canaglifolzin, another promising SGLT2i, was assessed for its cardiovascular safety and efficacy in patients with T2D and high CV risks in the CANVAS program which integrates both CANVAS and CANVAS-Renal trials. Observed effects on both CV and renal outcomes were comparable to EMPA-REG trial outcomes, yet with a difference in the degree of influence. Of note, heart failure patients treated with canagliflozin showed a lower risk of hospitalization with no statistical significance [33]. Subgroup analysis of EMPA-REG Outcome trial revealed similar but significant findings by showing that Empa addition to standard care in T2D patients and high CVD risks reduced heart failure hospitalization and cardiovascular death to the same extent in the presence or absence of heart failure at baseline when compared to their relative placebo group. However, rate of hospitalization per 1000 patients per years was lower in patients without heart failure at baseline when compared to patients with heart failure at baseline [34]. Whether SGLT2i CV benefits is a class effect remains to be determined by the outcome of ongoing trials examining the effects of other SGLT2 inhibitor molecules and expected to report in 2019 [25].
EMPA-REG Outcome Trial Summary |
Description and general outcomes A multicenter, randomized, double-blind, placebo-controlled trial that aims to assess the CV protective effect of Empa in patients with T2D associated to a high risk for CV events. ➢ Compared to placebo, Empa showed better glycemic control and advanced management of T2D’s deleterious effects on CV events (mortality among T2D patients with CVD). Study characteristics • 63.1 years old mean aged, 7028 patients; • Empa 10 mg (n = 2345), 25 mg (n = 2342) per day • Matching placebo (n = 2333) • Enrollees characteristics: White 72%, Asian 22%, other 6% • 57 % diagnosed with T2D > 10 years: • History of MI: 47% multivessel disease and 47% CAD • Antidiabetic therapy unchanged for 12 weeks prior to randomization Inclusion criteria • T2D with established CVD • Age ≥ 18 years old • HbA1c of ≥ 7.0% and ≤ 10% for patients on background therapy or HbA1c ≥ 7.0% and ≤ 9.0% for drug-naive patients • BMI ≤ 45 kg/m2 • GFR > 30 ml/min/1.73 m2 General endpoints outcome¥ ➢ 14% reduction in major CV events , 38% reductions in CV mortality, 35% reduction in heart failure hospitalization, 32% reduction of death from any cause. Primary outcomes ↓ CV death (3.7 vs. 5.9%, p < 0.001) ↓ All MI (4.8 vs. 5.4%, p = 0.23) ↓ All stroke ( 3.5 vs. 3.0%, p = 0.26) Secondary outcomes: ↓ Preload, afterload burden to heart, SBP, DBP ↓ Arterial stiffness ↓ CHF hospitalization or CV death( 5.7 vs. 8.5%, p < 0.001) ↓ Coronary revascularization(7 vs. 8%, p = 0.11) ↓ BW, BV ↓ Oxidative stress ↓ Visceral adiposity ↓ Hyperinsulinemia ↓ HbA1c, albuminuria ↓ Uric acid level ↑ Glycosuria, fasting, and postmeal glucagon concentration ↑ Hematocrit (5% in absolute values, and 11% in percentage points) ↑ Ketonemia, natriuresis and osmotic diuresis |
Nevertheless, considering that a significant proportion of T2D patients already show evidence of microvascular complications at the time of initial diagnosis of diabetes [35], a thorough examination of the underlying mechanism of SGLT2 inhibition-mediated cardioprotective effect is warranted. Identification of the target(s) for this “pleiotropic effect” is required to develop an understanding of their potential benefit and guide further research on rational and justified use in patients. Herein, we will review some potential molecular mechanisms through which SGLT2i can facilitate this cardioprotective effect (Fig. 1).

SGLT2 inhibitors impact on cardiac homeostasis following systemic and direct myocardial effects. Together SGLT2 mediated systemic and direct myocardial effects potentiate cardioprotective outcomes in T2D patients. Ang1-7, angiotensin 1-7; HBA1c, glycosylated hemoglobin; hs-CRP, high sensitive C reactive protein; NaCl, sodium chloride; FFA, free fatty acids; SGLT, sodium/glucose cotransporter; T2D, type 2 diabetes; CV, cardiovascular; Ca2+, calcium; Na2+, sodium; NHE, cardiac Na+/H+ exchanger; PP1, protein phosphatase 1; PLB, phospholamban; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca(2+)ATPase 2a; [Ca2+]c, cardiac cytoplasmic Ca2+ concentration; M1, M1 macrophages; M2, M2 macrophage; RONS, reactive oxygen and nitrogen species; NLRP-3 inflammasome, nucleotide-binding domain leucine-rich repeat containing protein inflammasome; AGEs, advanced glycoxidation end products; TG, triglyceride; FA, fatty acids; SNS, sympathetic nervous system;
The diabetic myocardium
Diabetes is associated with 2- to 4-fold increase in the risk for cardiovascular disease [36,37,38]. Seventy five to 80% of the deaths in patients with T2D are associated with a thrombotic event and ~ 70% of diabetic people > 65 years of age will die of some form of heart disease [37]. Prevalence of T2D or impaired glucose tolerance may be as high as 65% in MI patients, increasing the risk of mortality and congestive heart failure development [39, 40]. Heart failure is preceded by metabolic and mitochondrial dysfunction, oxidative stress, and cardiac myocytes death that are exacerbated in T2D patients with no defined mechanisms [36, 38]. To date, multiple studies emphasize the positive correlation between acute hyperglycemia and detrimental cardiac remodeling and prognosis post-MI [41,42,43,44,45,46]. Elevated plasma glucose on admission post-MI is a powerful prognostic tool for both in-hospital and long-term outcome in both diabetic and non-diabetic patients. In fact, there is positive correlation between plasma glucose level and mortality level post-MI, although the basis for the harmful effect of hyperglycemia is not understood [43, 44, 47]. A 4% increase in mortality is encountered for every 18 mg/dL increase in plasma glucose level [46]. Patients with and without established diabetes have comparable mortality rate when post-MI admission glucose levels are more than 200 mg/dL suggesting an acute glucose-mediated toxicity on the myocardium [46].
Mechanisms behind worsened cardiac remodeling post-injury in diabetic hearts remain unclear [48]. Hearts of diabetic patients are associated with contractile and relaxation dysfunction as well as an increased arrhythmia risk that are linked to autonomic neuropathy and sympatho-parasympathetic imbalance caused by parasympathetic denervation and sympathetic hyperinnervation [49,50,51]. In addition to the autonomic dysfunction, ion homeostasis and electrophysiological properties of the diabetic myocardium at the tissue level are also altered [52,53,54,55]. Metabolic aberrations, oxidative damage, and inflammation are also attributed to cardiac dysfunction in diabetic patients and further discussed in this review.
SGLT2 inhibitors and ion homeostasis of the diabetic myocardium
Myocardial Ca2+ and Na+ homeostasis is critical for proper cardiac signal transduction, heart rhythm regulation, and cardiomyocyte energy production and respiration [56, 57]. Rapid and proper change of intracellular Ca2+ concentration is essential for cardiac myocytes contraction and relaxation and is normally regulated by ion exchangers and channels including L-type Ca2+ channels, ryanodine receptor, Na+/Ca2+ exchanger (NCX), and sarcoplasmic-reticulum calcium ATPase 2a (SERCA2a) [56,57,58]. Na+ homeostasis on the other hand is regulated mainly by NCX, Na+/H+ exchanger (NHE), and the Na+/K+ pump and directly affects myocardial Ca2+ dynamics [52, 59]. Both Ca2+ and Na+ transport, handling and regulation are altered in the diabetic myocardium [52,53,54,55]. Altered Na+ transport in diabetic heart is attributed to decreased Na+/K+ pump and NCX activities but enhanced NHE activity overloading the cytosol with Na+ [55, 60,61,62,63,64]. Recent findings suggest an important role of SGLTi on ion homeostasis in T2D heart and a potential protective impact on T2D cardiac remodeling (Table 1). In a recent study, Lambert et al. found increased SGLT1 expression in failing hearts of T2D patients compared to controls, and linked Na+ overload in T2D rat myocytes to increased SGLT1-mediated Na+/glucose uptake [53]. Using dapagliflozin (Dapa), a selective SGLT2i, Hamouda et al. tested the electromechanical function of isolated ventricular myocytes of streptozotocin (STZ)-induced diabetic rats [73]. Their findings revealed a reduction in ventricular myocyte shortening and the amplitude of the intracellular Ca2+ transients in both STZ and control myocytes with greater effect in STZ myocytes, 5 min after Dapa exposure. The exact mechanism behind the negative inotropic effects is unclear but most probably is linked to alteration in the mechanisms of Ca2+ transport since together the myofilament sensitivity to Ca2+ and sarcoplasmic reticulum Ca2+ release were not altered by Dapa in both groups [73]. Indeed, in a very recent study, Baartscheer and colleagues confirmed the ion concentration alteration hypothesis [72]. Using Empa, another selective SGLT2i, ion homeostasis was tested on isolated ventricular myocytes from rabbits and rats in the presence of increased levels of extracellular glucose. Findings revealed that Empa decreased cardiac myocytes cytosolic Na+ [Na+]c and cytosolic Ca2+ [Ca2+]c levels and increased myocytes’ mitochondrial Ca2+ concentration by modulating NHE activity [72]. Although SGLT2 expression is not found in neither healthy nor pathological heart tissue [9, 75], Empa impact on ion homeostasis in cardiomyocytes has been linked to its potential direct interaction with NHE [72]. Data supporting this notion have been generated by using Cariporide, a well-known specific NHE inhibitor. Of note Cariporide attenuates intracellular Na+ accumulation during ischemia and Ca2+ accumulation during both ischemia and reperfusion, mechanisms that are proven to be cardioprotective by limiting infarct expansion and border-zone extension [76,77,78]. Cardioprotective impact of Cariporide was confirmed clinically in patients with acute ischemic coronary event undergoing PTCA or CABG procedures attenuating adverse remodeling within both the infarcted and non-infarcted areas of the heart [76, 79,80,81,82]. Application of Cariporide in the presence of Empa had minimal effect on Empa-induced cytosolic Na+ reduction and vice versa. Additionally, recovery of pH following acute acidic load was inhibited in the presence of Cariporide but strongly reduced with Empa [72]. These findings reinforce the potential direct NHE inhibition of Empa which is yet to be fully elucidated. In summary, decreasing intracellular Na+ levels by inhibiting SGLT2 and attenuating NHE activity results in increased Ca2+ uptake into the mitochondria and efflux into the extracellular space probably through NCX activity, decreasing intracellular Ca2+ levels and subsequently, improving calcium handling between cardiac cycles. These direct effects on the myocardium correlate well with the reduction in sympathetic activity observed in SGLT2i-treated T2D patients and support the cardioprotective effects of SGLT2i in T2D-heart failure patients [83,84,85]. On a functional level, two studies recently emerged supporting diastolic function improvement in T2D mouse model following SGLT2 inhibition [70, 71]. In the first study, Empa treatment in T2D ob/ob mouse model showed an increase in the myocardium contractile reserve following dobutamine stress challenge along with an increase in calcium handling through enhancing SERCA2a activity and improving left ventricular (LV) maximum pressure and diastolic function parameters [70]. In T2D female db/db mouse model exposed to Empa treatment, diastolic function significantly improved as evidenced by decreased LV filling pressure and enhanced septal wall motion, all in absence of any changes in BP [71]. In addition to Ca2+ handling improvement, diastolic function enhancement was linked to the antifibrotic aspect of Empa treatment. Serum and glucocorticoid-regulated kinase 1 (SGK1)/Enac profibrotic signaling pathway and the associated myocardial interstitial fibrosis decreased in the presence of Empa [71]. Notably, SGK1 is highly expressed and activated in the diabetic heart in the presence of an excess of circulating glucose and is directly linked to cardiac pro-fibrotic/hypertrophic effects [86,87,88]
SGLT2 inhibitors and metabolic alteration of the diabetic myocardium
Under physiologic conditions, 95% of myocardial energy is supplied via mitochondrial oxidative metabolism. Free fatty acids (FFAs), glucose, lactate, ketone bodies, and amino acids are all involved in oxidative metabolism. However, most of the energy production is obtained from FFAs and glucose metabolism with a negligible contribution of other substrates [89]. Myocardial metabolism can change depending on cardiac stress, substrate availability, and hormonal situation. Lipotoxicity, glucotoxicity, ketone bodies oxidation, and mitochondrial dysfunction are all associated with metabolic abnormalities of the diabetic myocardium. With T2D, insulin resistance increases lipolysis and subsequent fatty acid (FA) uptake and triglyceride (TG) storage into the myocardium [90]. Consequently, myocardial FA abundance amplifies the reliance on β-oxidation and FA storage while inhibiting pyruvate dehydrogenase (PDH) through PPARα-mediated PDK4 activation and subsequently inhibiting glucose oxidation, increasing the risk of myocardial steatosis and cytotoxicity [91,92,93,94,95]. Cardiac steatosis is considered a powerful predictor of cardiac dysfunction and cardiac remodeling and highly correlates with obese and T2D patients [96,97,98,99,100,101]. Inhibiting cardiac glucose oxidation raises cardiomyocytes glucose levels along with the risk of protein glycation and the formation of advanced glycation end-products (AGEs). Multiple metabolic pathways including pentose phosphate pathway (PPP), hexosamine biosynthesis pathway (HBP), and glycogeneic pathways are altered in this process, affecting the myocardium negatively [102,103,104,105]. AGEs formation is associated with cellular dysfunction via reactive oxygen species (ROS) production, and cross-linking with multiple macromolecules including SERCA, collagen, and ryanodine receptor leading to ventricular stiffness and dysfunction [106, 107]. LV dysfunction in T2D patients and rodents has also been linked to mitochondrial dysfunction [108,109,110,111]. Reduction in oxidative phosphorylation (OxPhos) limits ATP supply to the myocardium leading to systolic and diastolic impairment [110, 112,113,114,115]. Decreased OxPhos rate and increased FA oxidation will also increase ROS production due to high electron leakage in the mitochondrial respiratory chain. As expected, excessive ROS production amplifies T2D-mediated cardiac remodeling by inducing acute cellular damage and inflammatory responses [116, 117]. To date, the impact of SGLT2 inhibition on cardiac metabolic impairment has not been thoroughly investigated nor deciphered (Table 1). In their study, Joubert at al. used a unique non-obese mouse model of T2D known as the lipodystrophic Bscl2−/− (seipin knockout [SKO]) mouse to investigate the impact of SGLT2i on glucotoxicity in the absence of lipotoxicity [69]. SKO mice are characterized by an excessive increase in myocardial glucose uptake without lipid accumulation or lipotoxic features. Using Dapa as SGLT2 inhibitor and pioglitazone as insulin sensitizer, data revealed a more pronounced cardioprotective effect of Dapa-treated group compared to pioglitazone-treated group despite similar glucose lowering effects of both drugs [69]. This study supports direct cardioprotective effects of SGLT2i independently of glycemic control. In another study, the impact of SGLT2i on lipotoxicity was tested in a high-fat-high-sugar (HFHS) mouse model [68]. HFHS animals displayed T2D characteristics including high lipid deposition in both heart and liver along with hyperglycemia and insulin resistance. In addition to glycemic control, Empa treatment in HFHS group significantly mitigated myocardial and liver steatosis by reducing TG accumulation. Although Empa treatment significantly decreased TG plasma level with no effect on diet-induced increase of plasma total cholesterol and HDL-cholesterol levels, it is not clear whether the observed Empa effect on cardiac TG accumulation is also tissue-specific and requires further investigation [68, 118]. Metabolically, one possible explanation for SGLT2 inhibition-mediated cardioprotective effects is ketone bodies formation [13]. Ketone bodies are generated through FA metabolism in the liver with low plasma concentration under physiologic conditions [119]. With diabetes however, low plasma insulin, insulin resistance, lipolysis, and subsequent high FA levels accelerate ketone bodies formation and their importance as energy source for myocardium increases [120]. Multiple experimental studies showed that β-hydroxybutyrate, a ketone body, competes with FFA and glucose entry into cardiac mitochondrial metabolic oxidation with higher energy efficiency and lower myocardial oxygen consumption [121,122,123]. Unlike FFA oxidation, β-hydroxybutyrate generates less ROS and possesses antioxidants capacities which maintain mitochondrial integrity [124]. Additionally, ketone bodies increase mitochondrial biogenesis and exert anti-arrhythmic effects by stabilizing cell membrane potential [125]. In diet-induced obese diabetic rats, treatment with SGTL2i promotes lipolysis instead of glucose oxidation as a source of energy [126, 127]. SGLT2 inhibition also increased plasma ketone bodies levels in both experimental and clinical T2D along with shifting substrate usage from carbohydrates to lipids [126,127,128,129,130,131]. In summary, there is no clear understanding of how SGLT2i exert their cardioprotective effects through myocardial metabolism modulation. Evidence supports a direct effect of SGLT2i on reducing plasma glucose levels and shifting myocardial metabolism to FA and ketone bodies oxidation along with appropriate non-accumulative myocardial FA storage. Glucose lowering effects of antidiabetic drugs by itself induces lipolysis and ketone body formation as a compensatory mechanism [132]. SGLT2 inhibition however, improves lipolysis and limits TG accumulation in the liver and the myocardium [68]. Additionally, recent evidence has emerged showing an improvement in myocardial insulin sensitivity and glucose utilization following Empa treatment in a T2D ob/ob mouse model [70]. Improvement of myocardial insulin sensitivity could be linked to the significant decrease in epicardial fat volume (EFV) that was observed in T2D patients treated with SGLT2i [133, 134]. Of note, EFV accumulation highly correlates with cardio-metabolic risks including insulin resistance and inflammation [135, 136]. All the above findings suggest that reducing plasma glucose levels, improving myocardial insulin sensitivity and glucose utilization, along with directly lowering TG accumulation in the myocardium could allow ketone bodies cardioprotective metabolism to dominate while limiting myocardial glucotoxic and lipotoxic effects. Investigating this concept with stronger evidence such as direct SGLT2 inhibiting effect on improving myocardial ketone bodies uptake and oxidation, increasing myocardial FA oxidation and subsequently lowering myocardial TG accumulation, and ultimately restoring pre-diabetic glucose-FA metabolic balance is warranted. However, a critical clinical complication known as euglycemic diabetic ketoacidosis (DKA) has emerged, not so infrequently, in individuals treated with SGLT2i. DKA has the potential of becoming a life-threatening condition due to systemic ketone bodies accumulation consequent to SGLT2 inhibition-dependent decrease in insulin secretion and subsequent increase in glucagon secretion and activation of lipolysis [137,138,139].
SGLT2 inhibition and oxidative inflammatory response with the diabetic cardiovascular system
Oxidative stress and chronic systemic inflammation are closely associated and long-known to play a key role in the pathogenesis of diabetes-induced CVD. They are crucial members of the vicious cycle of diabetes, which also includes hyperglycemia, insulin resistance, and dyslipidemia [140,141,142,143,144]. When it comes to oxidative stress and inflammation in the diabetic myocardium, investigations take into consideration the micro and macrovascular complications of diabetes and the direct impact on end-organ damage. Endothelial function by itself is vital for proper homeostasis of the body and its dysfunction is directly associated with multiple pathophysiological abnormalities including acute coronary syndrome and cardiomyopathy [145]. In the diabetic myocardium, oxidative stress plays a major role in promoting cardiac inflammation and fibrosis [146,147,148]. In fact, several studies have documented a significant reduction in cardiac pro-inflammatory and fibrotic markers upon treatment with antioxidants [143, 147, 149]. Conclusively, microvascular, macrovascular, and cardiac dysfunction with diabetes could not be evaluated as independent entities since they are functionally interconnected and directly affected by systemic oxidative stress and inflammation.
Effects on the myocardium
The impact of SGLT2 inhibition on the myocardial oxidative and inflammatory response has been investigated in multiple studies (Table 1). In a prediabetic rat model of metabolic syndrome, 10 weeks of treatment with Empa significantly reduced cardiomyocytes hypertrophy, interstitial fibrosis, and subcutaneous fat tissue despite no significant change in BP and autonomic function [67]. Reduction of cardiac oxidative stress and inflammation following Empa accounted for the observed direct cardioprotective effects [67]. In a T2D mouse model, administration of Empa to db/db mice for a 10-week period significantly improved cardiac and pericoronary arterial fibrosis, and myocardial macrophage infiltration with no impact on BP [66]. Findings were directly linked to significant reduction in cardiac superoxide production supporting the antioxidant capacities of SGLT2is [66]. In an attempt to better understand the impact of SGLT2 inhibition on cardiac inflammatory modulation, Dapa was tested on a MI rat model. Dapa administration over a period of 4 weeks post-MI resulted in significant decrease in reactive oxygen and nitrogen species (RONS) as early as 3 days post-MI followed by a significant decrease in myofibroblast infiltration and cardiac fibrosis, 28 days post-MI [65]. Those results were attributed to an enhanced M2 macrophage polarization and IL-10 anti-inflammatory cytokine upregulation through a RONS-attenuation-mediated STAT3 activation signaling pathway [65]. Of note, antioxidants are known to increase STAT3 activity during MI and to polarize, along with STAT3 activation, macrophages towards an M2 anti-inflammatory phenotype [150,151,152,153], supporting the role of SGLT2i as antioxidant and inflammatory modulators in the heart. Modulation of cardiac inflammation by SGLT2i was directly tested on nucleotide-binding domain leucine-rich repeat containing protein (NLRP)-3 inflammasome activation [154]. NLRP-3 inflammasome is an intracellular oligomer, which promotes the activation of the pro-inflammatory cytokines IL-1β and IL-18 and is directly involved in the pathogenesis of some metabolic disorders including obesity induced insulin resistance, diabetes mellitus, and atherosclerosis [154,155,156,157]. In T2D models, NLRP-3 inflammasome is upregulated in the myocardium and contribute to cardiac inflammation, fibrosis, and subsequent cardiac dysfunction and cardiomyopathy [158,159,160,161,162]. Dapa treatment in T2D ob/ob mice significantly reduced cardiac NLRP-3 inflammasome activation along with antifibrotic effects and overall improvement in LV ejection fraction and systolic function when compared to controls [163]. Observed non-functional effects were replicated in vitro, ruling out any glucose or hemodynamic-lowering dependent effects [163].
Effects on the vasculature
Early experiments with phlorizin, a natural product with non-selective SGLT inhibitory properties, revealed that inhibition of glucose entry into vascular smooth muscle cells via this transporter modulated both intracellular calcium levels and serotonin-mediated contractions [164]. Using phlorizin, SGLT was postulated to act as a glucose sensor in retinal pericytes whereby capillary tone and microvascular blood flow would be regulated based on extracellular glucose levels [165]. Attenuation of smooth muscle contraction by SGLT blockade was reported in lymphatic preparations as well and was linked to smooth muscle ion homeostasis [166]. Increasing vascular tone was attributed to the sodium component of the SGLT transport process driven by glucose, whereby sodium entering the cell would later be exchanged for extracellular calcium via NCX activity and hence, the increased vascular tone [166]. A better-studied aspect of the effect of SGLT function on vascular tone is through its action on endothelial cells. Several lines of evidence implicated an enhanced SGLT activity in vascular endothelial cells under stress conditions [167]. SGLT inhibition by phlorizin ameliorated hypoxia-induced endothelial cell activation and production of vasoactive prostaglandins and platelet-activating factors [168]. Indeed, simulation of stroke conditions in vitro led to an increase in SGLT-mediated glucose transport, which upon inhibition in in vivo models of stroke was associated with a reduced brain infarct size and edema [169]. With regard to endothelium-dependent relaxation, acute exposure to glucose and insulin was associated with endothelial NO production attributed to sodium entry via SGLT and later exchange with extracellular calcium [170]. It followed that SGLT inhibition with phlorizin strongly attenuated endothelium-dependent NO-dependent vasodilation ex vivo [170]. Such an observation is contradictory to a proposed vasculoprotective effect for SGLT inhibitors in diabetic vascular disorders of which endothelium dysfunction is considered a hallmark. However, observed in vitro results with phlorizin are questionable for three major reasons: (1) phlorizin is not specific to SGLT and has broad off-target effects; (2) phlorizin could be hydrolyzed either chemically (as may occur in aqueous solutions) or enzymatically to phloretin, which inhibits most passive glucose transporters (GLUTs) and not just the SGLTs [171]; and (3) in vitro studies focus on acute and limited rather than chronic and systemic effects of the drugs. Chronic and systemic effects are essential in multifactorial diseases such as diabetes. A newer study examined the effect of chronic in vivo phlorizin treatment (10 weeks) on a diabetic mouse model [172]. Isolated aortas from the treated mice showed an improved endothelium-dependent relaxation compared to the untreated diabetic animals. This was attributed to an apparent reduction in oxidative stress and AGEs rather than direct vasoactive effects as hypothesized in acute in vitro studies. In an attempt to confirm these findings with more selective SGLT inhibitor drugs, a study compared ex vivo and in vivo treatment effects using chronic phlorizin and canagliflozin, a highly specific SGLT2 inhibitor, on mouse coronary arteries [74]. The authors found that, despite the observation that both SGLT inhibitors did not affect NO-dependent coronary relaxation ex vivo, arteries isolated from diabetic animals receiving 4-week canagliflozin treatment had improved relaxation compared to untreated diabetic cohorts. Of note though, in the latter study, the authors were not able to detect SGLT2 expression in mouse or human coronary vascular smooth muscle and endothelia to justify canagliflozin vasoactive action, which question the direct mechanisms behind SGLT2 inhibition on the vasculature. Recent studies with Empa and ipragliflozin, both novel selective and highly specific SGLT2i, fortified these findings [66, 173]. Empa significantly improved pericoronary arterial fibrosis, coronary arterial thickening, and vasodilation impairment following a 10-week treatment in T2D db/db mice. In this model, Empa protective effects were attributed to significant attenuation of oxidative stress in cardiovascular tissue [66]. Ipragliflozin was tested in STZ-induced diabetic mouse model on a 3-week administration timeframe. Findings of the ipragliflozin study revealed a protection against endothelial dysfunction via modulation of inflammation and attenuation of oxidative stress [173]. Taken together, the previous results indicate that the potential vasculoprotection associated with SGLT2i therapy is mediated by an indirect modulatory effect, and most probably through attenuating oxidative stress and inflammation, thus supporting their capacity in reversing the perivascular adipose inflammation associated with insulin resistance and diabetes [174, 175].
A significant body of literature underscores the role of inflammation in the vasculature and perivascular adipose tissue in diabetic vasculopathy [174]. A vascular anti-inflammatory role for SGLT2i is supported by recent evidence whereby the beneficial vascular effect of chronic oral SGLT2 inhibitor treatment was accompanied by a reduced vascular expression of inflammatory molecules, e.g., monocyte chemoattractant protein-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule [173]. Dapa treatment was shown to decrease both visceral and sub-cutaneous adipose tissue mass in diabetic patients along with the decrease in high sensitivity C-reactive protein (hsCRP) serum levels, a well-known marker of inflammation in CVD [176, 177]. Nevertheless, SGLT2 inhibitor-mediated vasculoprotective effect through suppression of perivascular adipose inflammation remains to be examined systematically.
Protective mechanisms of SGLT2 inhibition in heart failure
Both, heart failure with preserved and reduced ejection fraction are debilitating complexed clinical syndromes that are often accompanied with comorbidities that directly affect patient’s prognosis and clinical intervention [178, 179]. The alteration of cardiac stroke volume in heart failure could be related to three major affected mechanisms: preload, afterload, and myocardial contractility. Clinically heart failure patients treated with SGLT2i exhibited a lower risk of hospitalization when compared to placebo [33, 34]. SGLT2 inhibition may decrease preload through promoting osmotic diuresis which reduces volume overload in heart failure patients improving myocardial stretching mechanisms and contractility [180]. Recent studies revealed a reduction in blood pressure, arterial stiffness, and vascular resistance in T2D patients treated with Empa, effects that could decrease afterload and subsequently improve cardiac output in heart failure patients [16]. In addition to the protective SGLT2 inhibition impact on multiple mechanisms preceding heart failure and diabetic cardiomyopathy development including metabolic impairment, mitochondrial dysfunction, calcium handling, inflammation, and oxidative stress, SGLT2i impact on BMI, a well-known heart failure risk factor, is also promising [13, 181, 182]. In fact, treatment with SGLT2 inhibitor results in approximately 2–3 kg reduction body weight over 24–52 weeks in diabetic patients most probably due to osmotic diuresis and caloric loss as well as other undefined mechanisms [13, 182, 183]. Although SGLT2i-mediated weight reduction is small, its combination with modest reduction in preload and afterload could synergistically improve cardiac workload and contractility [184].
Conclusions and future perspectives
To date, no antidiabetic drug showed the same CV benefit as SGLT2is did with diabetic patients. Although other anti-diabetic drugs appeared to produce CV benefit when given to pre-diabetic patients, e.g., metformin [185] and pioglitazone [186], neither drug showed the same benefit in diabetic patients. Clinical data describing the cardiovascular benefits of SGLT2is in diabetic patients highlight the potential these drugs offer as future therapeutic tools to address cardiovascular deterioration in an impaired metabolic milieu. Recent studies showed that SGLT1, but not SGLT2, is found in the capillaries and myocytes of human and rodent hearts and upregulated in the myocardium of multiple pathological conditions including T2D, hypertrophy, heart failure, and infarcted hearts [9,10,11,12, 75]. SGLT1 effect in heart remains controversial as it has been directly linked to NOX2-mediated ROS production in cardiomyocytes but also shown to play a critical role in cardiac cell protection during the acute phase of ischemia-reperfusion injury by regulating cardiac energy metabolism [187,188,189]. These findings question whether the direct observed SGLT2 inhibition effects on the CV system are mediated through off-target effects such as SGLT1 upregulation or inhibition. To date, no data support the upregulation of SGLT1 in the heart following SGLT2 inhibition as a compensatory mechanism similarly to what is found in the kidneys and GI tract [190]. Although canagliflozin expressed a modest SGLT-1 inhibitory effect in the small intestine from the luminal side at clinical dosage, it did not affect SGLT-1 in the heart or the skeletal muscle [191]. Nonetheless, there is a consensus that SGLT2i direct effects on the myocardium (Table 1) are independent of SGLT2i-mediated systemic effects (Table 2), and together direct and systemic effects potentiate SGLT2 inhibition cardioprotective outcomes (Fig. 1). Several questions remain to be answered in terms of the exact mechanism of action upon chronic use, optimal timing of intervention, and whether structural and functional modifications of these molecules could serve to enhance the CV protective effect and thus, maximizing their therapeutic benefit. Finally, exploring the effects of these drugs in pre-diabetic or non-diabetic individuals with other forms of metabolic impairment and at high risk of CVD is also warranted.
Abbreviations
- SGLT:
-
sodium/glucose cotransporter
- WHO:
-
World Health Organization
- Ca2+ :
-
calcium
- T2D:
-
type 2 diabetes
- CV:
-
cardiovascular
- Empa:
-
empagliflozin
- MI:
-
myocardial infarction
- CVD:
-
cardiovascular disease
- NHE:
-
sodium hydrogen exchanger
- NCX:
-
sodium calcium exchanger
- SERCA2a:
-
sarcoplasmic-reticulum calcium ATPase 2a
- Dapa:
-
dapagliflozin
- STZ:
-
streptozotocin
- [Na+]c:
-
cytoplasmic Na+ concentration
- [Ca2+]c:
-
cytoplasmic Ca2+ concentration
- ob/ob:
-
leptin-deficient homozygous T2D mouse model
- LV:
-
left ventricle
- db/db:
-
C57BLKS/J-Leprdb/Leprdb T2D mouse model
- SGK1:
-
serum/glucocorticoid regulated kinase 1
- FFAs:
-
free fatty acids
- TG:
-
triglyceride
- PDH:
-
pyruvate dehydrogenase
- PPARα:
-
peroxisome proliferator-activated receptor alpha
- PDK4:
-
pyruvate dehydrogenase lipoamide kinase isozyme 4
- AGEs:
-
advanced glycation end products
- PPP:
-
pentose phosphate pathway
- HBP:
-
hexosamine biosynthesis pathway
- ROS:
-
reactive oxygen species
- OxPhos:
-
oxidative phosphorylation
- ATP:
-
adenosine triphosphate
- SKO:
-
lipodystrophic Bscl2−/− (seipin knockout [SKO]) T2D mouse model
- HFHS:
-
high-fat-high-sugar
- EFV:
-
epicardial fat volume
- DKA:
-
diabetic ketoacidosis
- RONS:
-
reactive oxygen and nitrogen species
- NLRP:
-
nucleotide binding domain leucine rich repeat containing protein
- IL:
-
interleukin
- NO:
-
nitric oxide
- Phlor:
-
phlorizin
- Cana:
-
canagliflozin
- hsCRP:
-
high sensitivity C-reactive protein
References
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15(7):539–553. https://doi.org/10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
Nathan DM (1993) Long-term complications of diabetes mellitus. N Engl J Med 328(23):1676–1685. https://doi.org/10.1056/NEJM199306103282306
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3(11):e442. https://doi.org/10.1371/journal.pmed.0030442
Cefalu WT, Leiter LA, Yoon KH, Arias P, Niskanen L, Xie J, Balis DA, Canovatchel W, Meininger G (2013) Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 382(9896):941–950. https://doi.org/10.1016/S0140-6736(13)60683-2
Forst T, Guthrie R, Goldenberg R, Yee J, Vijapurkar U, Meininger G, Stein P (2014) Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes on background metformin and pioglitazone. Diabetes Obes Metab 16(5):467–477. https://doi.org/10.1111/dom.12273
Purnell JQ, Weyer C (2003) Weight effect of current and experimental drugs for diabetes mellitus: from promotion to alleviation of obesity. Treat Endocrinol 2(1):33–47
Tahrani AA, Bailey CJ, Del Prato S, Barnett AH (2011) Management of type 2 diabetes: new and future developments in treatment. Lancet 378(9786):182–197. https://doi.org/10.1016/S0140-6736(11)60207-9
Fala L (2015) Jardiance (empagliflozin), an SGLT2 inhibitor, receives FDA approval for the treatment of patients with type 2 diabetes. Am Health Drug Benefits 8(Spec Feature):92–95
Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, Radovic N, Jadrijevic S, Aleksic I, Walles T, Sauvant C, Sabolic I, Koepsell H (2015) Localizations of Na(+)-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467(9):1881–1898. https://doi.org/10.1007/s00424-014-1619-7
Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F (2009) SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res 84(1):111–118. https://doi.org/10.1093/cvr/cvp190
Elfeber K, Stumpel F, Gorboulev V, Mattig S, Deussen A, Kaissling B, Koepsell H (2004) Na(+)-D-glucose cotransporter in muscle capillaries increases glucose permeability. Biochem Biophys Res Commun 314(2):301–305
Zhou L, Cryan EV, D'Andrea MR, Belkowski S, Conway BR, Demarest KT (2003) Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem 90(2):339–346. https://doi.org/10.1002/jcb.10631
Ferrannini E, Mark M, Mayoux E (2016) CV protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care 39(7):1108–1114. https://doi.org/10.2337/dc16-0330
Cherney DZ, Perkins BA, Soleymanlou N, Har R, Fagan N, Johansen OE, Woerle HJ, von Eynatten M, Broedl UC (2014) The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc Diabetol 13:28. https://doi.org/10.1186/1475-2840-13-28
Scheerer MF, Rist R, Proske O, Meng A, Kostev K (2016) Changes in HbA1c, body weight, and systolic blood pressure in type 2 diabetes patients initiating dapagliflozin therapy: a primary care database study. Diabetes Metab Syndr Obes 9:337–345. https://doi.org/10.2147/dmso.s116243
Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, Johansen OE (2015) Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab 17(12):1180–1193. https://doi.org/10.1111/dom.12572
Ott C, Jumar A, Striepe K, Friedrich S, Karg MV, Bramlage P, Schmieder RE (2017) A randomised study of the impact of the SGLT2 inhibitor dapagliflozin on microvascular and macrovascular. Circulation. 16(1):26. https://doi.org/10.1186/s12933-017-0510-1
Daneman D (2006) Type 1 diabetes. Lancet 367(9513):847–858. https://doi.org/10.1016/S0140-6736(06)68341-4
Disease GBD, Injury I, Prevalence C (2016) Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388(10053):1545–1602. https://doi.org/10.1016/S0140-6736(16)31678-6
Leiter LA, Yoon K-H, Arias P, Langslet G, Xie J, Balis DA, Millington D, Vercruysse F, Canovatchel W, Meininger G (2015) Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care 38(3):355–364. https://doi.org/10.2337/dc13-2762
Matthaei S, Bowering K, Rohwedder K, Grohl A, Parikh S (2015) Dapagliflozin improves glycemic control and reduces body weight as add-on therapy to metformin plus sulfonylurea: a 24-week randomized, double-blind clinical trial. Diabetes Care 38(3):365–372. https://doi.org/10.2337/dc14-0666
Cefalu WT, Riddle MC (2015) SGLT2 inhibitors: the latest “new kids on the block”! Diabetes Care 38(3):352–354. https://doi.org/10.2337/dc14-3048
Kitada M, Zhang Z, Mima A, King GL (2010) Molecular mechanisms of diabetic vascular complications. J Diabetes Investig 1(3):77–89. https://doi.org/10.1111/j.2040-1124.2010.00018.x
American_Diabetes_Association (2016) Standards of medical care in diabetes-2016: glycemic targets. Diabetes Care 39(Supplement 1):S39–S46. https://doi.org/10.2337/dc16-S008
Holman RR, Sourij H, Califf RM (2014) Cardiovascular outcome trials of glucose-lowering drugs or strategies in type 2 diabetes. Lancet 383(9933):2008–2017. https://doi.org/10.1016/S0140-6736(14)60794-7
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW (2008) 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359(15):1577–1589. https://doi.org/10.1056/NEJMoa0806470
Jax TW (2010) Metabolic memory: a vascular perspective. Cardiovasc Diabetol 9:51. https://doi.org/10.1186/1475-2840-9-51
Basnet S, Kozikowski A, Makaryus AN, Pekmezaris R, Zeltser R, Akerman M, Lesser M, Wolf-Klein G (2015) Metformin and myocardial injury in patients with diabetes and ST-segment elevation myocardial infarction: a propensity score matched analysis. J Am Heart Assoc 4(10):e002314. https://doi.org/10.1161/JAHA.115.002314
Lexis CP, van der Horst IC, Lipsic E, Wieringa WG, de Boer RA, van den Heuvel AF, van der Werf HW, Schurer RA, Pundziute G, Tan ES, Nieuwland W, Willemsen HM, Dorhout B, Molmans BH, van der Horst-Schrivers AN, Wolffenbuttel BH, ter Horst GJ, van Rossum AC, Tijssen JG, Hillege HL, de Smet BJ, van der Harst P, van Veldhuisen DJ, Investigators G-I (2014) Effect of metformin on left ventricular function after acute myocardial infarction in patients without diabetes: the GIPS-III randomized clinical trial. JAMA 311(15):1526–1535. https://doi.org/10.1001/jama.2014.3315
Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB (2016) Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 375(4):311–322. https://doi.org/10.1056/NEJMoa1603827
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373(22):2117–2128. https://doi.org/10.1056/NEJMoa1504720
Tikkanen I, Narko K, Zeller C, Green A, Salsali A, Broedl UC, Woerle HJ (2015) Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care 38(3):420–428. https://doi.org/10.2337/dc14-1096
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, Group CPC (2017) Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377(7):644–657. https://doi.org/10.1056/NEJMoa1611925
Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, investigators E-ROt (2016) Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur Heart J 37(19):1526–1534. https://doi.org/10.1093/eurheartj/ehv728
Fowler MJ (2008) Microvascular and macrovascular complications of diabetes. Clin Diabetes 26(2):77–82. https://doi.org/10.2337/diaclin.26.2.77
Kannel WB, McGee DL (1979) Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care 2(2):120–126
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S (2016) Executive summary: heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133(4):447–454. https://doi.org/10.1161/CIR.0000000000000366
Stamler J, Vaccaro O, Neaton JD, Wentworth D (1993) Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 16(2):434–444
Ishihara M (2012) Acute hyperglycemia in patients with acute myocardial infarction. Circ J 76(3):563–571
Oswald GA, Corcoran S, Yudkin JS (1984) Prevalence and risks of hyperglycaemia and undiagnosed diabetes in patients with acute myocardial infarction. Lancet 1(8389):1264–1267
Cid-Alvarez B, Gude F, Cadarso-Suarez C, Gonzalez-Babarro E, Rodriguez-Alvarez MX, Garcia-Acuna JM, Gonzalez-Juanatey JR (2009) Admission and fasting plasma glucose for estimating risk of death of diabetic and nondiabetic patients with acute coronary syndrome: nonlinearity of hazard ratios and time-dependent comparison. Am Heart J 158(6):989–997. https://doi.org/10.1016/j.ahj.2009.10.004
Dirkali A, van der Ploeg T, Nangrahary M, Cornel JH, Umans VA (2007) The impact of admission plasma glucose on long-term mortality after STEMI and NSTEMI myocardial infarction. Int J Cardiol 121(2):215–217. https://doi.org/10.1016/j.ijcard.2006.08.107
Capes SE, Hunt D, Malmberg K, Gerstein HC (2000) Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 355(9206):773–778. https://doi.org/10.1016/S0140-6736(99)08415-9
Malmberg K, Norhammar A, Wedel H, Ryden L (1999) Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation 99(20):2626–2632
Dziewierz A, Giszterowicz D, Siudak Z, Rakowski T, Dubiel JS, Dudek D (2010) Admission glucose level and in-hospital outcomes in diabetic and non-diabetic patients with acute myocardial infarction. Clin Res Cardiol 99(11):715–721. https://doi.org/10.1007/s00392-010-0175-1
Stranders I, Diamant M, van Gelder RE, Spruijt HJ, Twisk JW, Heine RJ, Visser FC (2004) Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus. Arch Intern Med 164(9):982–988. https://doi.org/10.1001/archinte.164.9.982
Wahab NN, Cowden EA, Pearce NJ, Gardner MJ, Merry H, Cox JL, Investigators I (2002) Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J Am Coll Cardiol 40(10):1748–1754
Lejay A, Fang F, John R, Van JA, Barr M, Thaveau F, Chakfe N, Geny B, Scholey JW (2016) Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J Mol Cell Cardiol 91:11–22. https://doi.org/10.1016/j.yjmcc.2015.12.020
Kuehl M, Stevens MJ (2012) Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat Rev Endocrinol 8(7):405–416. https://doi.org/10.1038/nrendo.2012.21
Vinik AI, Maser RE, Ziegler D (2011) Autonomic imbalance: prophet of doom or scope for hope? Diabet Med 28(6):643–651. https://doi.org/10.1111/j.1464-5491.2010.03184.x
Xuan YL, Wang Y, Xue M, Hu HS, Cheng WJ, Li XR, Yin J, Yang N, Yan SH (2015) In rats the duration of diabetes influences its impact on cardiac autonomic innervations and electrophysiology. Auton Neurosci 189:31–36. https://doi.org/10.1016/j.autneu.2015.01.003
Despa S, Bers DM (2013) Na(+) transport in the normal and failing heart—remember the balance. J Mol Cell Cardiol 61:2–10. https://doi.org/10.1016/j.yjmcc.2013.04.011
Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S (2015) Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc 4(9):e002183. https://doi.org/10.1161/JAHA.115.002183
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57(4):660–671. https://doi.org/10.1007/s00125-014-3171-6
Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando S, Kemmotsu O, Kanno M (2000) Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J Physiol 527(Pt 1):85–94
Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR, Xie ZJ (2015) Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol 593(6):1361–1382. https://doi.org/10.1113/jphysiol.2014.282319
Kho C, Lee A, Hajjar RJ (2012) Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat Rev Cardiol 9(12):717–733. https://doi.org/10.1038/nrcardio.2012.145
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868):198–205. https://doi.org/10.1038/415198a
Cingolani HE, Ennis IL (2007) Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 115(9):1090–1100. https://doi.org/10.1161/CIRCULATIONAHA.106.626929
Anzawa R, Bernard M, Tamareille S, Baetz D, Confort-Gouny S, Gascard JP, Cozzone P, Feuvray D (2006) Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia 49(3):598–606. https://doi.org/10.1007/s00125-005-0091-5
Chattou S, Diacono J, Feuvray D (1999) Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol Scand 166(2):137–144. https://doi.org/10.1046/j.1365-201x.1999.00547.x
Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D (2007) Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia 50(6):1335–1344. https://doi.org/10.1007/s00125-007-0628-x
Hansen PS, Clarke RJ, Buhagiar KA, Hamilton E, Garcia A, White C, Rasmussen HH (2007) Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol 292(3):C1070–C1077. https://doi.org/10.1152/ajpcell.00288.2006
Kjeldsen K, Braendgaard H, Sidenius P, Larsen JS, Norgaard A (1987) Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes 36(7):842–848
Lee TM, Chang NC, Lin SZ (2017) Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med 104:298–310. https://doi.org/10.1016/j.freeradbiomed.2017.01.035
Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K, Ma M, Nakagawa T, Kusaka H, Kim-Mitsuyama S (2014) Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol 13:148. https://doi.org/10.1186/s12933-014-0148-1
Kusaka H, Koibuchi N, Hasegawa Y, Ogawa H, Kim-Mitsuyama S (2016) Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc Diabetol 15(1):157. https://doi.org/10.1186/s12933-016-0473-7
Benetti E, Mastrocola R, Vitarelli G, Cutrin JC, Nigro D, Chiazza F, Mayoux E, Collino M, Fantozzi R (2016) Empagliflozin protects against diet-induced NLRP-3 inflammasome activation and lipid accumulation. J Pharmac Exp Ther 359(1):45–53. https://doi.org/10.1124/jpet.116.235069
Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, Dollet L, Le May C, Toumaniantz G, Manrique A, Charpentier F, Staels B, Magre J, Cariou B, Prieur X (2017) The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes 66(4):1030–1040. https://doi.org/10.2337/db16-0733
Hammoudi N, Jeong D, Singh R, Farhat A, Komajda M, Mayoux E, Hajjar R, Lebeche D (2017) Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Ther. https://doi.org/10.1007/s10557-017-6734-1
Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, Barron B, Mayoux E, Rector RS, Whaley-Connell A, DeMarco VG (2017) Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol 16(1):9. https://doi.org/10.1186/s12933-016-0489-z
Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ (2017) Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 60(3):568–573. https://doi.org/10.1007/s00125-016-4134-x
Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC (2015) Dapagliflozin reduces the amplitude of shortening and Ca(2+) transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem 400(1-2):57–68. https://doi.org/10.1007/s11010-014-2262-5
Han Y, Cho YE, Ayon R, Guo R, Youssef KD, Pan M, Dai A, Yuan JX, Makino A (2015) SGLT inhibitors attenuate NO-dependent vascular relaxation in the pulmonary artery but not in the coronary artery. Am J Physiol Lung Cell Mol Physiol 309(9):L1027–L1036. https://doi.org/10.1152/ajplung.00167.2015
Di Franco A, Cantini G, Tani A, Coppini R, Zecchi-Orlandini S, Raimondi L, Luconi M, Mannucci E (2017) Sodium-dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol 243:86–90. https://doi.org/10.1016/j.ijcard.2017.05.032
Avkiran M, Cook AR, Cuello F (2008) Targeting Na+/H+ exchanger regulation for cardiac protection: a RSKy approach? Curr Opin Pharmacol 8(2):133–140. https://doi.org/10.1016/j.coph.2007.12.007
Gumina RJ, Mizumura T, Beier N, Schelling P, Schultz JJ, Gross GJ (1998) A new sodium/hydrogen exchange inhibitor, EMD 85131, limits infarct size in dogs when administered before or after coronary artery occlusion. J Pharmac Exp Ther 286(1):175–183
Rohmann S, Weygandt H, Minck KO (1995) Preischaemic as well as postischaemic application of a Na+/H+ exchange inhibitor reduces infarct size in pigs. Cardiovasc Res 30(6):945–951
Bolli R (2003) The role of sodium-hydrogen ion exchange in patients undergoing coronary artery bypass grafting. J Card Surg 18(Suppl 1):21–26
Boyce SW, Bartels C, Bolli R, Chaitman B, Chen JC, Chi E, Jessel A, Kereiakes D, Knight J, Thulin L, Theroux P, Investigators GDIANS (2003) Impact of sodium-hydrogen exchange inhibition by cariporide on death or myocardial infarction in high-risk CABG surgery patients: results of the CABG surgery cohort of the GUARDIAN study. J Thorac Cardiovasc Surg 126(2):420–427
Rupprecht HJ, vom Dahl J, Terres W, Seyfarth KM, Richardt G, Schultheibeta HP, Buerke M, Sheehan FH, Drexler H (2000) Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation 101(25):2902–2908
Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, White HD, Willerson JT, Jessel A (2000) Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation 102(25):3032–3038
Sano M (2017) Hemodynamic effects of sodium-glucose cotransporter 2 inhibitors. J Clin Med Res 9(6):457–460. https://doi.org/10.14740/jocmr3011w
Martens P, Mathieu C, Verbrugge FH (2017) Promise of SGLT2 inhibitors in heart failure: diabetes and beyond. Curr Treat Options Cardiovasc Med 19(3):23. https://doi.org/10.1007/s11936-017-0522-x
Rahman A, Hitomi H, Nishiyama A (2017) Cardioprotective effects of SGLT2 inhibitors are possibly associated with normalization of the circadian rhythm of blood pressure. Hypertens Res. https://doi.org/10.1038/hr.2016.193
Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A (2005) Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation 111(13):1652–1659. https://doi.org/10.1161/01.CIR.0000160352.58142.06
Das S, Aiba T, Rosenberg M, Hessler K, Xiao C, Quintero PA, Ottaviano FG, Knight AC, Graham EL, Bostrom P, Morissette MR, del Monte F, Begley MJ, Cantley LC, Ellinor PT, Tomaselli GF, Rosenzweig A (2012) Pathological role of serum- and glucocorticoid-regulated kinase 1 in adverse ventricular remodeling. Circulation 126(18):2208–2219. https://doi.org/10.1161/CIRCULATIONAHA.112.115592
Lang F, Shumilina E (2013) Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27(1):3–12. https://doi.org/10.1096/fj.12-218230
Mudaliar S, Alloju S, Henry RR (2016) Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care 39(7):1115–1122. https://doi.org/10.2337/dc16-0542
Labbe SM, Grenier-Larouche T, Noll C, Phoenix S, Guerin B, Turcotte EE, Carpentier AC (2012) Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 61(11):2701–2710. https://doi.org/10.2337/db11-1805
Ferre P (2004) The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes 53(Suppl 1):S43–S50
Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, Bax JJ, de Roos A, Twisk JW, Heine RJ, Lammertsma AA, Smit JW, Diamant M (2009) Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol 54(16):1524–1532. https://doi.org/10.1016/j.jacc.2009.04.074
Huang B, Wu P, Bowker-Kinley MM, Harris RA (2002) Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 51(2):276–283
Wieland O, Siess E, Schulze-Wethmar FH, von Funcke HG, Winton B (1971) Active and inactive forms of pyruvate dehydrogenase in rat heart and kidney: effect of diabetes, fasting, and refeeding on pyruvate dehydrogenase interconversion. Arch Biochem Biophys 143(2):593–601
Wu P, Peters JM, Harris RA (2001) Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun 287(2):391–396. https://doi.org/10.1006/bbrc.2001.5608
Glenn DJ, Wang F, Nishimoto M, Cruz MC, Uchida Y, Holleran WM, Zhang Y, Yeghiazarians Y, Gardner DG (2011) A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension 57(2):216–222. https://doi.org/10.1161/HYPERTENSIONAHA.110.160655
Oka T, Topper YJ (1972) Dynamics of insulin action on mammary epithelium. Nat New Biol 239(94):216–217
Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ (2008) Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52(22):1793–1799. https://doi.org/10.1016/j.jacc.2008.07.062
Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, Unger R, Victor RG (2003) Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med 49(3):417–423. https://doi.org/10.1002/mrm.10372
Szczepaniak LS, Victor RG, Orci L, Unger RH (2007) Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res 101(8):759–767. https://doi.org/10.1161/CIRCRESAHA.107.160457
Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 97(4):1784–1789
Chung SS, Ho EC, Lam KS, Chung SK (2003) Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 14(8 Suppl 3):S233–S236
Laughlin MR, Petit WA Jr, Shulman RG, Barrett EJ (1990) Measurement of myocardial glycogen synthesis in diabetic and fasted rats. Am J Physiol 258(1 Pt 1):E184–E190
McNulty PH (2007) Hexosamine biosynthetic pathway flux and cardiomyopathy in type 2 diabetes mellitus. Focus on “Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart.”. Am J Physiol Cell Physiol 292(4):C1243–C1244. https://doi.org/10.1152/ajpcell.00521.2006
Sochor M, Gonzalez AM, McLean P (1984) Regulation of alternative pathways of glucose metabolism in rat heart in alloxan diabetes: changes in the pentose phosphate pathway. Biochem Biophys Res Commun 118(1):110–116
Brownlee M (1995) Advanced protein glycosylation in diabetes and aging. Annu Rev Med 46:223–234. https://doi.org/10.1146/annurev.med.46.1.223
Petrova R, Yamamoto Y, Muraki K, Yonekura H, Sakurai S, Watanabe T, Li H, Takeuchi M, Makita Z, Kato I, Takasawa S, Okamoto H, Imaizumi Y, Yamamoto H (2002) Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol 34(10):1425–1431
Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD (2009) Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol 54(20):1891–1898. https://doi.org/10.1016/j.jacc.2009.07.031
Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, Nichols CE, Long DM, Olfert IM, Jagannathan R, Hollander JM (2014) Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol 307(1):H54–H65. https://doi.org/10.1152/ajpheart.00845.2013
Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B (2014) Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130(7):554–564. https://doi.org/10.1161/CIRCULATIONAHA.113.008476
Zorzano A, Liesa M, Palacin M (2009) Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 41(10):1846–1854. https://doi.org/10.1016/j.biocel.2009.02.004
Diamant M, Lamb HJ, Groeneveld Y, Endert EL, Smit JW, Bax JJ, Romijn JA, de Roos A, Radder JK (2003) Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J Am Coll Cardiol 42(2):328–335
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70(2):200–214
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779):787–790. https://doi.org/10.1038/35008121
Savabi F, Kirsch A (1991) Alteration of the phosphocreatine energy shuttle components in diabetic rat heart. J Mol Cell Cardiol 23(11):1323–1333
Ballinger SW, Patterson C, Yan CN, Doan R, Burow DL, Young CG, Yakes FM, Van Houten B, Ballinger CA, Freeman BA, Runge MS (2000) Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res 86(9):960–966
Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD (2004) Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India 52:794–804
Mazidi M, Rezaie P, Gao HK, Kengne AP (2017) Effect of sodium-glucose cotransport-2 inhibitors on blood pressure in people with type 2 diabetes mellitus: a systematic review and meta-analysis of 43 randomized control trials with 22 528 patients. J Am Heart Assoc 6(6). https://doi.org/10.1161/JAHA.116.004007
Balasse EO, Fery F (1989) Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 5(3):247–270
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85(3):1093–1129. https://doi.org/10.1152/physrev.00006.2004
Kashiwaya Y, King MT, Veech RL (1997) Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. Am J Cardiol 80(3A):50A–64A
Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, Chance B, Clarke K, Veech RL (1995) Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J 9(8):651–658
Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD (2003) beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol 285(4):H1626–H1631. https://doi.org/10.1152/ajpheart.00332.2003
Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E (2013) Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339(6116):211–214. https://doi.org/10.1126/science.1227166
Cotter DG, Schugar RC, Crawford PA (2013) Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol 304(8):H1060–H1076. https://doi.org/10.1152/ajpheart.00646.2012
Suzuki M, Takeda M, Kito A, Fukazawa M, Yata T, Yamamoto M, Nagata T, Fukuzawa T, Yamane M, Honda K, Suzuki Y, Kawabe Y (2014) Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr Diabetes 4:e125. https://doi.org/10.1038/nutd.2014.20
Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, Miyoshi S, Tahara A, Kurosaki E, Li Q, Tomiyama H, Sasamata M, Shibasaki M, Uchiyama Y (2014) SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol 727:66–74. https://doi.org/10.1016/j.ejphar.2014.01.040
Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA (2012) Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity (Silver Spring) 20(8):1645–1652. https://doi.org/10.1038/oby.2012.59
Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR, Muscelli E (2016) Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 65(5):1190–1195. https://doi.org/10.2337/db15-1356
Inagaki N, Kondo K, Yoshinari T, Takahashi N, Susuta Y, Kuki H (2014) Efficacy and safety of canagliflozin monotherapy in Japanese patients with type 2 diabetes inadequately controlled with diet and exercise: a 24-week, randomized, double-blind, placebo-controlled, phase III study. Expert Opin Pharmacother 15(11):1501–1515. https://doi.org/10.1517/14656566.2014.935764
Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, Broedl UC, Woerle HJ (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124(2):499–508. https://doi.org/10.1172/JCI72227
Fukao T, Lopaschuk GD, Mitchell GA (2004) Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 70(3):243–251. https://doi.org/10.1016/j.plefa.2003.11.001
Bouchi R, Terashima M, Sasahara Y, Asakawa M, Fukuda T, Takeuchi T, Nakano Y, Murakami M, Minami I, Izumiyama H, Hashimoto K, Yoshimoto T, Ogawa Y (2017) Luseogliflozin reduces epicardial fat accumulation in patients with type 2 diabetes: a pilot study. Cardiovasc Diabetol 16(1):32. https://doi.org/10.1186/s12933-017-0516-8
Fukuda T, Bouchi R, Terashima M, Sasahara Y, Asakawa M, Takeuchi T, Nakano Y, Murakami M, Minami I, Izumiyama H, Hashimoto K, Yoshimoto T, Ogawa Y (2017) Ipragliflozin reduces epicardial fat accumulation in non-obese type 2 diabetic patients with visceral obesity: a pilot study. Diabetes Ther. https://doi.org/10.1007/s13300-017-0279-y
Iacobellis G, Leonetti F (2005) Epicardial adipose tissue and insulin resistance in obese subjects. J Clin Endocrinol Metab 90(11):6300–6302. https://doi.org/10.1210/jc.2005-1087
Rijzewijk LJ, Jonker JT, van der Meer RW, Lubberink M, de Jong HW, Romijn JA, Bax JJ, de Roos A, Heine RJ, Twisk JW, Windhorst AD, Lammertsma AA, Smit JW, Diamant M, Lamb HJ (2010) Effects of hepatic triglyceride content on myocardial metabolism in type 2 diabetes. J Am Coll Cardiol 56(3):225–233. https://doi.org/10.1016/j.jacc.2010.02.049
Kibbey RG (2015) SGLT-2 inhibition and glucagon: Cause for alarm? Trends Endocrinol Metab 26(7):337–338. https://doi.org/10.1016/j.tem.2015.05.011
Ogawa W, Sakaguchi K (2016) Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: possible mechanism and contributing factors. J Diabetes Investig 7(2):135–138. https://doi.org/10.1111/jdi.12401
Peters AL, Buschur EO, Buse JB, Cohan P, Diner JC, Hirsch IB (2015) Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care 38(9):1687–1693. https://doi.org/10.2337/dc15-0843
Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, Bufe A, Barroso MC, Lankisch M (2009) Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol 8:58. https://doi.org/10.1186/1475-2840-8-58
Haffner SM (2006) The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol 97(2A):3A–11A. https://doi.org/10.1016/j.amjcard.2005.11.010
Khullar M, Al-Shudiefat AA, Ludke A, Binepal G, Singal PK (2010) Oxidative stress: a key contributor to diabetic cardiomyopathy. Can J Physiol Pharmacol 88(3):233–240. https://doi.org/10.1139/Y10-016
Rochette L, Zeller M, Cottin Y, Vergely C (2014) Diabetes, oxidative stress and therapeutic strategies. Biochim Biophys Acta 1840(9):2709–2729. https://doi.org/10.1016/j.bbagen.2014.05.017
Paoletti R, Bolego C, Poli A, Cignarella A (2006) Metabolic syndrome, inflammation and atherosclerosis. Vasc Health Risk Manag 2(2):145–152
Sena CM, Pereira AM, Seica R (2013) Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta 1832(12):2216–2231. https://doi.org/10.1016/j.bbadis.2013.08.006
Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM (2006) Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 20(9):1546–1548. https://doi.org/10.1096/fj.05-4642fje
Suzuki H, Kayama Y, Sakamoto M, Iuchi H, Shimizu I, Yoshino T, Katoh D, Nagoshi T, Tojo K, Minamino T, Yoshimura M, Utsunomiya K (2015) Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 64(2):618–630. https://doi.org/10.2337/db13-1896
Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y (2008) Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem 317(1-2):43–50. https://doi.org/10.1007/s11010-008-9803-8
Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, Ogawa H, Kim-Mitsuyama S (2010) Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens 28(2):340–352. https://doi.org/10.1097/HJH.0b013e32833366cd
Jadhav A, Tiwari S, Lee P, Ndisang JF (2013) The heme oxygenase system selectively enhances the anti-inflammatory macrophage-M2 phenotype, reduces pericardial adiposity, and ameliorated cardiac injury in diabetic cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther 345(2):239–249. https://doi.org/10.1124/jpet.112.200808
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, Lei S, Liu Z, Ng KF, Wong GT, Vanhoutte PM, Irwin MG, Xia Z (2013) N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med 63:291–303. https://doi.org/10.1016/j.freeradbiomed.2013.05.043
Shiraishi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M (2012) Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun 425(2):304–308. https://doi.org/10.1016/j.bbrc.2012.07.086
Sica A, Bronte V (2007) Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 117(5):1155–1166. https://doi.org/10.1172/JCI31422
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10(2):417–426
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735. https://doi.org/10.1146/annurev-immunol-031210-101405
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464(7293):1357–1361. https://doi.org/10.1038/nature08938
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17(2):179–188. https://doi.org/10.1038/nm.2279
Fuentes-Antras J, Ioan AM, Tunon J, Egido J, Lorenzo O (2014) Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol 2014:847827. https://doi.org/10.1155/2014/847827
Luo B, Li B, Wang W, Liu X, Liu X, Xia Y, Zhang C, Zhang Y, Zhang M, An F (2014) Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther 28(1):33–43. https://doi.org/10.1007/s10557-013-6498-1
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, Zhang M, Zhang Y, An F (2014) NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PloS One 9(8):e104771. https://doi.org/10.1371/journal.pone.0104771
Shah MS, Brownlee M (2016) Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res 118(11):1808–1829. https://doi.org/10.1161/CIRCRESAHA.116.306923
Singh LP (2014) The NLRP3 inflammasome and diabetic cardiomyopathy : editorial to: “Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model” by Beibei Luo et al. Cardiovasc Drugs Ther 28(1):5–6. https://doi.org/10.1007/s10557-013-6501-x
Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y (2017) SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther 31(2):119–132. https://doi.org/10.1007/s10557-017-6725-2
Kahn AM, Lichtenberg RA, Allen JC, Seidel CL, Song T (1995) Insulin-stimulated glucose transport inhibits Ca2+ influx and contraction in vascular smooth muscle. Circulation 92(6):1597–1603
Wakisaka M, Kitazono T, Kato M, Nakamura U, Yoshioka M, Uchizono Y, Yoshinari M (2001) Sodium-coupled glucose transporter as a functional glucose sensor of retinal microvascular circulation. Circ Res 88(11):1183–1188
Li X, Mizuno R, Ono N, Ohhashi T (2008) Glucose and glucose transporters regulate lymphatic pump activity through activation of the mitochondrial ATP-sensitive K+ channel. J Physiol Sci 58(4):249–261. https://doi.org/10.2170/physiolsci.RP004608
Nishizaki T, Matsuoka T (1998) Low glucose enhances Na+/glucose transport in bovine brain artery endothelial cells. Stroke 29(4):844–849
Berna N, Arnould T, Remacle J, Michiels C (2001) Hypoxia-induced increase in intracellular calcium concentration in endothelial cells: role of the Na(+)-glucose cotransporter. J Cell Biochem 84(1):115–131
Vemula S, Roder KE, Yang T, Bhat GJ, Thekkumkara TJ, Abbruscato TJ (2009) A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J Pharmacol Exp Ther 328(2):487–495. https://doi.org/10.1124/jpet.108.146589
Taubert D, Rosenkranz A, Berkels R, Roesen R, Schomig E (2004) Acute effects of glucose and insulin on vascular endothelium. Diabetologia 47(12):2059–2071. https://doi.org/10.1007/s00125-004-1586-1
Mudaliar S, Polidori D, Zambrowicz B, Henry RR (2015) Sodium-glucose cotransporter inhibitors: effects on renal and intestinal glucose transport: from bench to bedside. Diabetes Care 38(12):2344–2353. https://doi.org/10.2337/dc15-0642
Shen L, You BA, Gao HQ, Li BY, Yu F, Pei F (2012) Effects of phlorizin on vascular complications in diabetes db/db mice. Chin Med J 125(20):3692–3696
Salim HM, Fukuda D, Yagi S, Soeki T, Shimabukuro M, Sata M (2016) Glycemic control with ipragliflozin, a novel selective SGLT2 inhibitor, ameliorated endothelial dysfunction in streptozotocin-induced diabetic mouse. Front Cardiovasc Med 3:43. https://doi.org/10.3389/fcvm.2016.00043
Lastra G, Manrique C (2015) Perivascular adipose tissue, inflammation and insulin resistance: link to vascular dysfunction and cardiovascular disease. Horm Mol Biol Clin Invest 22(1):19–26. https://doi.org/10.1515/hmbci-2015-0010
Roma-Lavisse C, Tagzirt M, Zawadzki C, Lorenzi R, Vincentelli A, Haulon S, Juthier F, Rauch A, Corseaux D, Staels B, Jude B, Belle EV, Susen S, Chinetti-Gbaguidi G, Dupont A (2015) M1 and M2 macrophage proteolytic and angiogenic profile analysis in atherosclerotic patients reveals a distinctive profile in type 2 diabetes. Diab Vasc Dis Res 12(4):279–289. https://doi.org/10.1177/1479164115582351
Bolinder J, Ljunggren Ö, Kullberg J, Johansson L, Wilding J, Langkilde AM, Sugg J, Parikh S (2012) Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 97(3):1020–1031. https://doi.org/10.1210/jc.2011-2260
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF (2010) Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care 33(10):2217–2224. https://doi.org/10.2337/dc10-0612
Borlaug BA (2014) The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 11(9):507–515. https://doi.org/10.1038/nrcardio.2014.83
Kemp CD, Conte JV (2012) The pathophysiology of heart failure. Cardiovasc Pathol 21(5):365–371. https://doi.org/10.1016/j.carpath.2011.11.007
Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Fagan NM, Woerle HJ, Johansen OE, Broedl UC, von Eynatten M (2014) Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129(5):587–597. https://doi.org/10.1161/CIRCULATIONAHA.113.005081
Clark AL, Fonarow GC, Horwich TB (2014) Obesity and the obesity paradox in heart failure. Prog Cardiovasc Dis 56(4):409–414. https://doi.org/10.1016/j.pcad.2013.10.004
Monica Reddy RP, Inzucchi SE (2016) SGLT2 inhibitors in the management of type 2 diabetes. Endocrine 53(2):364–372. https://doi.org/10.1007/s12020-016-0943-4
Abdul-Ghani M, Del Prato S, Chilton R, DeFronzo RA (2016) SGLT2 inhibitors and cardiovascular risk: lessons learned from the EMPA-REG OUTCOME study. Diabetes Care 39(5):717–725. https://doi.org/10.2337/dc16-0041
Pham SV, Chilton R (2017) EMPA-REG OUTCOME: the cardiologist’s point of view. Am J Med 130(6S):S57–S62. https://doi.org/10.1016/j.amjmed.2017.04.006
Diabetes Prevention Program Research G (2015) Long-term effects of lifestyle intervention or metformin on diabetes development and microvascular complications over 15-year follow-up: the Diabetes Prevention Program Outcomes Study. Lancet Diabetes Endocrinol 3(11):866–875. https://doi.org/10.1016/S2213-8587(15)00291-0
Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R, Brass LM, Schwartz GG, Adams HP Jr, Berger L, Carolei A, Clark W, Coull B, Ford GA, Kleindorfer D, O'Leary JR, Parsons MW, Ringleb P, Sen S, Spence JD, Tanne D, Wang D, Winder TR, Investigators IT (2016) Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 374(14):1321–1331. https://doi.org/10.1056/NEJMoa1506930
Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C (2011) NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res 92(2):237–246. https://doi.org/10.1093/cvr/cvr230
Kanwal A, Nizami HL, Mallapudi S, Putcha UK, Mohan GK, Banerjee SK (2016) Inhibition of SGLT1 abrogates preconditioning-induced cardioprotection against ischemia-reperfusion injury. Biochem Biophys Res Commun 472(2):392–398. https://doi.org/10.1016/j.bbrc.2016.02.016
Kashiwagi Y, Nagoshi T, Yoshino T, Tanaka TD, Ito K, Harada T, Takahashi H, Ikegami M, Anzawa R, Yoshimura M (2015) Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice. PloS One 10(6):e0130605. https://doi.org/10.1371/journal.pone.0130605
Liu JJ, Lee T, DeFronzo RA (2012) Why Do SGLT2 inhibitors inhibit only 30-50% of renal glucose reabsorption in humans? Diabetes 61(9):2199–2204. https://doi.org/10.2337/db12-0052
Ohgaki R, Wei L, Yamada K, Hara T, Kuriyama C, Okuda S, Ueta K, Shiotani M, Nagamori S, Kanai Y (2016) Interaction of the sodium/glucose cotransporter (SGLT) 2 inhibitor canagliflozin with SGLT1 and SGLT2. J Pharmacol Exp Ther 358(1):94–102. https://doi.org/10.1124/jpet.116.232025
Acknowledgements
The authors are grateful for the generous support of the Department of Pharmacology and Toxicology of the University of Mississippi Medical Center and the Department of Pharmacology and Toxicology at the American University of Beirut.
Funding Source
This work was supported by grants from the American University of Beirut (Seed grant #100410, MPP grant #320145) to FAZ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors confirm that there are no conflicts of interest or disclosure to make.
Rights and permissions
About this article

Cite this article
Kaplan, A., Abidi, E., El-Yazbi, A. et al. Direct cardiovascular impact of SGLT2 inhibitors: mechanisms and effects. Heart Fail Rev 23, 419–437 (2018). https://doi.org/10.1007/s10741-017-9665-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-017-9665-9