
Abstract
The highly regulated processes of mitochondrial fusion (joining), fission (division) and trafficking, collectively called mitochondrial dynamics, determine cell-type specific morphology, intracellular distribution and activity of these critical organelles. Mitochondria are critical for cardiac function, while their structural and functional abnormalities contribute to several common cardiovascular diseases, including heart failure (HF). The tightly balanced mitochondrial fusion and fission determine number, morphology and activity of these multifunctional organelles. Although the intracellular architecture of mature cardiomyocytes greatly restricts mitochondrial dynamics, this process occurs in the adult human heart. Fusion and fission modulate multiple mitochondrial functions, ranging from energy and reactive oxygen species production to Ca2+ homeostasis and cell death, allowing the heart to respond properly to body demands. Tightly controlled balance between fusion and fission is of utmost importance in the high energy-demanding cardiomyocytes. A shift toward fission leads to mitochondrial fragmentation, while a shift toward fusion results in the formation of enlarged mitochondria and in the fusion of damaged mitochondria with healthy organelles. Mfn1, Mfn2 and OPA1 constitute the core machinery promoting mitochondrial fusion, whereas Drp1, Fis1, Mff and MiD49/51 are the core components of fission machinery. Growing evidence suggests that fusion/fission factors in adult cardiomyocytes play essential noncanonical roles in cardiac development, Ca2+ signaling, mitochondrial quality control and cell death. Impairment of this complex circuit causes cardiomyocyte dysfunction and death contributing to heart injury culminating in HF. Pharmacological targeting of components of this intricate network may be a novel therapeutic modality for HF treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In mammals, the relentlessly beating heart is one of the most mitochondria-enriched organs. Mitochondrial oxidative phosphorylation (OXPHOS) generates up to 90 % of ATP, required for constant contraction of cardiomyocytes, and the organelles occupy almost 30 % of their volume [1–3]. Over the past two decades, mitochondria have emerged not only as cellular powerhouses, but also as critical integrators of fundamental cellular processes, ranging from the generation of reactive oxygen species (ROS) and signal transduction to the maintenance of Ca2+ homeostasis, stress responses and cell death [4–6]. Comprehensive studies of cardiac mitochondria have convincingly demonstrated that their dysfunction is implicated in the pathogenesis of common cardiovascular diseases (CVD), such as dysrhythmias, myocardial ischemia, cardiomyopathies culminating in end-stage heart failure (HF) [7–10].
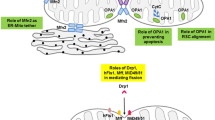
Mitochondria are dynamic organelles—the two opposing highly regulated processes, fusion (joining) and fission (division)—determine cell-type specific mitochondrial morphology, intracellular distribution and activity (Fig. 1). Furthermore, mitochondria can move along the cytoskeleton interacting with various intracellular organelles and ensuring region-specific cellular requirements. These finely tuned processes, which have been termed mitochondrial dynamics, modulate mitochondrial shape and function allowing living cells to respond properly to frequently changing environmental conditions [11–14].

Roles of mitochondrial dynamics. Red Mitochondria with high membrane potential, with high oxidative phosphorylation (OXPHOS) activity. Blue Mitochondria with low membrane potential. Mitofusin 1 or 2 (Mfn1, Mfn2) mediates mitochondrial outer-membrane fusion in a tissue-specific manner, and optic atrophy gene1 (OPA1) mediates inner-membrane fusion. The zinc metalloprotease OMA, also known as “Overlapping with the m-AAA protease 1 homolog,” is an essential enzyme in mitochondrial maintenance that proteolytically cleaves OPA1 under low membrane potential conditions, promoting fission. Mitochondrial dynamics factors 49 and 51 or mitochondrial fission factor (Mff) recruit dynamin-related protein 1 (DRP1) onto mitochondria at sites marked by endoplasmic reticulum tubules (ER), and DRP1 mediates mitochondrial division. In cultured cells, upon a decrease in mitochondrial membrane potential, PINK1 kinase recruits Parkin, a ubiquitin E3 ligase, which ubiquitinates several mitochondrial targets, including MFN1 and mitochondria Rho (Miro), to facilitate the degradation of mitochondria via mitophagy. Parkin-mediated ubiquitination triggers outer mitochondrial membrane-associated degradation (OMMAD)—a proteosomal pathway that degrades ubiquitinated OM proteins in a CDC48-dependent manner. OMMAD is probably cell type dependent and may also function in quality control. In erythrocytes, mitophagy receptor Nix1 is involved in autophagosome recruitment. ER forms close contacts with mitochondria, essential for calcium regulation in cellular microcompartments. Miro (blue feet) is a mitochondrial receptor for kinesin via Milton that facilitates the transport of mitochondria on microtubules in a Ca2+-regulated manner. Upon synaptic activity in neurons, influx of glutamate and Ca2+ halts mitochondrial transport via Miro to position them at sites of synaptic activity that require Ca2+ uptake and ATP. From Nunnari and Suomalainen [6] with permission of Elsevier
Mitochondrial fusion produces interconnected mitochondrial network and is essential for the maintenance and inheritance of mitochondrial DNA (mtDNA), the transmission of membrane potential and Ca2+ signaling along the mitochondrial network [12, 15, 16]. The opposing process, mitochondrial fission, leads to smaller, more discrete organelles and plays important roles in mitochondrial partitioning during mitosis, cytoskeleton-mediated trafficking to energy-demanding intracellular compartments and in selective autophagic removal of damaged mitochondria by the process called mitophagy [12, 15–17]. Moreover, elongation of mitochondrial tubules has been shown upon differentiation of progenitor cells into cardiomyocytes, while mitochondrial fragmentation can contribute to cytochrome c release leading eventually to apoptosis [18–21]. Alterations in the fine-tuned balance between mitochondrial fusion and fission are implicated in the pathogenesis of cancer, neurodegenerative, metabolic and cardiac disorders [15, 20, 22–25].
The mechanisms of mitochondrial dynamics have been studied mainly in cell types other than cardiomyocytes. The high energy need to fuel excitation–contraction coupling determines not only the great density of mitochondria, but also their specific arrangement within the cardiomyocytes. In mature cardiomyocytes, mitochondria are tightly packed between the sarcomere myofibrils or between the myofibrils and the plasma membrane, or clustered nearby the nucleus [1]. Furthermore, mitochondria in cardiomyocytes are closely associated with the sarcoplasmic reticulum (SR), the major compartment for Ca2+ storage and release, required for cardiac contraction [26, 27]. Such mitochondrial localization provides close contact with sarcomeres and efficient SR-mitochondrial crosstalk, linking high Ca2+ microdomains and energy generation organelles during excitation–contraction coupling [28]. However, this unique arrangement significantly restricts mitochondrial dynamics in adult cardiomyocytes compared to other cell types (e.g., neurons, fibroblasts or liver cells). Notably, the major proteins, which mediate mitochondrial dynamics, are highly expressed in the mammalian myocardium and their cardiomyocyte-specific genetic ablation is lethal [29–33]. Increasing evidence suggests that fusion/fission factors in adult cardiomyocytes play essential noncanonical roles in cardiac development, Ca2+ signaling, mitochondrial quality control and cell death.
In this review, we will provide an overview of the general mechanisms of mitochondrial fusion and fission, and the core proteins that mediate these complex processes. Then, we will discuss recent progress in our understanding of the role of mitochondrial dynamics in the pathogenesis of HF, focusing on noncanonical functions of fusion/fission proteins in the mitophagic removal of damaged mitochondria and in the initiation of cell death. The potential of therapeutic targeting of mitochondrial dynamics proteins will also be discussed.
Mechanisms of mitochondrial dynamics
Evolutionary conserved large GTPases, related to the dynamin superfamily, along with a number of binding partners promote both mitochondrial fusion and fission (Table 1) [20, 34–36]. Importantly, these dynamin family GTPases are highly expressed in the human adult heart.
Mitochondrial fusion
In mammals, the dynamin family GTPases—two mitofusin isoforms, Mfn1 (Mgm1 in yeast) and Mfn2, and optic atrophy protein 1 (OPA1; Fzo1 in yeast)—are the core components of the mitochondrial fusion machinery [20, 37].
Mfn1 and Mfn2 share a similar molecular architecture: the N-terminal GTPase domain, heptad-repeat domain 1 (HR1), two transmembrane (TM) domains, which anchor the proteins in the outer mitochondrial membrane (OMM), a short loop exposed in the intermembrane space (IMS) and the C-terminal heptad-repeat domain 2 (HR2) (Fig. 2a) [38]. The TM domains of Mfn1 and Mfn2 are embedded in the OMM, while their HR1 and HR2 protrude into the cytosol, where HR2 mediates interaction with their counterparts in adjacent mitochondria [38–40]. Mfn2, but not Mfn1, is also localized in the endoplasmic reticulum (ER)/SR and is involved in tethering of mitochondria with these organelles [41].

Molecular structure of core mitochondrial fusion and fission proteins. a Fusion proteins. Mitofusins, Mfn1, Mfn2, located in the outer mitochondrial membrane (OMM), contain a GTPase domain (GTPase), two heptad-repeat regions (HR1 and HR2) and two transmembrane (TM) domains. Phosphorylation sites of Mfn2 are also shown. OPA1 is located in the inner mitochondrial membrane (IMM) and contains mitochondrial targeting sequence (MTS), TM domain, heptad-repeat region (HR), GTPase domain, middle domain (MD) and GTPase effector domain (GED). Proteolytic cleavage within MTS by matrix-processing peptidase (MPP) and at the S1 and S2 sites by metalloproteases, which produce long OPA1 isoforms (L-OPA1) and short isoforms (S-OPA1), is shown by arrows. b Fission proteins. Cytoplasmic dynamin-related protein 1 (Drp1) contains a GTPase domain, middle domain (MD), GTPase effector domain (GED) and heptad-repeat region (HR). Phosphorylation sites of Drp1 are also depicted. Fission protein 1 (Fis1) and mitochondrial fission factor (Mff) are anchored by their TM domains to the OMM. In addition, Fis1 contains tetratricopeptide repeat (TPR), while Mff contains heptad-repeat region (HR) and coiled-coil region (CC)
OPA1 is also the dynamin-related GTPase composed of an N-terminal mitochondrial targeting sequence, cleaved by matrix-processing peptidase (MPP), TM domain, HR domain, GTPase domain, middle domain and a C-terminal GTPase effector domain (Fig. 2b) [42]. OPA1 is localized in the inner mitochondrial membrane (IMM) and the intermembrane space [43].
OPA1 is regulated at both mRNA and protein levels. Differential splicing generates eight distinct mRNA OPA1 splice forms depending on the tissue [44]. Multiple OPA1 isoforms result from processing at two sites between the N-terminal TM region and HR [20, 36]. In mammals, several proteases, such as presenilin-associated rhomboid-like protease (PARL), i-AAA metalloprotease (Ymel), m-AAA metalloprotease (paraplegin) and zinc metalloprotease OMA1, catalyze OPA1 cleavage [45–52]. As a result, long OPA1 isoforms (L-OPA1), containing the TM domain, and short isoforms (S-OPA1), lacking the TM domain, can be generated (Fig. 2b). L-OPA1 is anchored to the IMM by its TM domain, while S-OPA, which lacks the TM, is targeted to the IMM via its association with the IMM-anchored L-OPA1 [46, 47]. The loss of mitochondrial membrane potential induces L-OPA1 cleavage leading to accumulation of S-OPA1 isoforms and inhibition of fusion and targeting mitochondria to mitophagy [48, 53]. Furthermore, proapoptotic stimuli induce OMA1-mediated OPA1 cleavage resulting in the formation of fusion inactive S-OPA1 isoforms [50].
According to current paradigm, mitochondrial fusion is a 3-step process, which includes OMM tethering followed by a highly coordinated OMM and IMM fusion. First, the HR2 of mitofusins of adjacent mitochondria interacts to form homodimers (Mfn1–Mfn1 or Mfn2–Mfn2) or more potent heterodimers (Mfn1–Mfn2) to tether approaching mitochondria [40]. This initial docking step brings the OMM of two mitochondria close together to initiate the OMM fusion. After fusion of OMM, OPA1 mediates IMM fusion [54]. GTP hydrolysis catalyzed by the GTPase activities of Mfn1/Mfn2 and OPA1 provides energy for these processes [36]. In yeast, an additional OMM protein Ugo1 coordinates the OMM and IMM fusion [55, 56]. In higher eukaryotes, no structural or functional Ugo1 equivalents have been found; hence, the precise mechanism underlying the coupling of the OMM and IMM fusion remains to be determined. It has been hypothesized that interactions between mitofusins and OPA1 may be a part of such coordination mechanism [57].
Mitochondrial fission
In mammals, mitochondrial fission is under control of a 80-kDa dynamin-related protein 1 (Drp1; Dnm1 in yeast), also known as dynamin-like protein 1 (Dlp1) [17, 20, 37, 58]. Drp1 is the dynamin family GTPase composed of an N-terminal GTPase domain, middle domain and a C-terminal GTPase effector domain essential for self-assembly (Fig. 2b) [59]. Drp1 is mainly a soluble cytosolic protein, but its subpool colocalizes with mitochondria at sites of future fission [60, 61]. It forms dimers/tetramers in the cytosol and higher-order structures upon interaction with membranes [62, 63]. Cytosolic Drp1 is recruited to mitochondria by interaction with OMM proteins, such as Fis1, Mff and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51, respectively) [64–67].
Fis1 is a small (~17 kDa) single-pass TM protein with a C-tail anchored in the OMM [64, 68, 69]. Its N-terminal multiple tetratricopeptide repeat motif, facing the cytosol, is thought to be involved in the recruitment of Drp1 to mitochondria (Fig. 2b) [70–73]. Mff contains N-terminal heptad repeats, coiled-coil domain and a C-terminal TM tail, which anchor it to the OMM (Fig. 2b) [74]. In contrast to the uniform Fis1 localization in the OMM, Mff mainly colocalizes with the Drp1 foci on the OMM during fission.
However, the specific roles of Fis1, Mff and MiD49/51 in the recruitment of Drp1 to mitochondria and Drp1-mediated fission are uncertain [17, 58]. Yeast Fis1 is essential for the recruitment of Dnm1 (yeast orthologue of mammalian Drp1) [75], in mammals; however, Fis1 appears to be dispensable for Drp1 recruitment [65]. A recent study has shown that Fis1, Mff and MiD49/51 contribute to Drp1 recruitment; however, they can serve as Drp1 receptors on the OMM independently of each other [67]. Furthermore, both Fis1 and Mff, beyond their role in Drp1 recruiting, appear to facilitate Drp1 assembly into spirals on the OMM during fission [67].
According to current paradigm, mitochondrial fission is coupled to the inhibition of the mitochondrial fusion machinery. Cytosolic Drp1 as small oligomers is recruited to mitochondria through interactions with several OMM proteins, including Fis1, Mff and MiD49/51. Drp1 oligomers polymerize into spiral structures around the mitochondria and form fission foci, which constrict and divide mitochondria in a GTP-dependent manner [17, 58]. Recently, it has been shown that MiD51 can stimulate the GTPase activity of Drp1 and therefore assist Drp1-mediated constriction [76, 77]. Several additional proteins, such as mitochondrial protein of 18 kDa (MTP18), ganglioside-induced differentiation-associated protein 1 (GDAP1), endophilin B1 (Endo B1) and leucine-rich repeat kinase 2 (LRRK2), may contribute to mitochondrial fission; however, their roles in the process remain to be determined [17, 58].
Regulation of mitochondrial dynamics
Opposing processes of mitochondrial fusion and fission are tightly regulated to maintain mitochondrial morphology and function in response to changing conditions [17, 58]. The core components of the mammalian machineries that promote mitochondrial dynamics represent main targets for complex regulatory mechanisms operating at multiple levels.
Complex regulation of OPA1 by mRNA and proteolytic processing has already been described (Fig. 2b). Mfn2 concentration is regulated at the transcriptional level. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), one of the critical regulators of mitochondrial biogenesis, upregulates Mfn2 expression in response to increased metabolic demand [78]. Importantly, downregulation of PGC-1α associated with reduced Mfn2 expression has been found in rats and in patients with pulmonary arterial hypertension that can contribute to the development of HF [79, 80].
Furthermore, several E3 ubiquitin-protein ligases, including anaphase-promoting complex (APC/C)CDH1, Huwe1 and Parkin, catalyze the ubiquitination of both Mfn1 and Mfn2 inhibiting their fusion activity as well as targeting them to degradation [81–87]. Stress-induced mitochondrial depolarization—the sign of mitochondrial damage—targets PTEN-induced putative kinase 1 (PINK1) and/or JNK to the OMM [82]. PINK1 selectively accumulated on dysfunctional mitochondria phosphorylates Mfn2 on Thr111 and Ser442 recruits and activates cytosolic E3 ubiquitin ligase Parkin to ubiquitinate Mfn2 directing thereby damaged mitochondria to mitophagy [85, 88–91]. PINK1/Parkin mediates the ubiquitination of Mfn1 and Mfn2 as well as other mitochondrial proteins, such as voltage-dependent anion channel 1 (VDAC1) and p62 (also known as sequestosome 1), targeting them to proteosomal degradation [85, 92, 93].
Overexpression of fission protein Drp1 does not induce mitochondrial fission suggesting that post-translational modifications, which affect its mitochondrial recruitment, GTPase activity or self-assembly ability, play an essential regulatory role [17, 58]. Phosphorylation of Drp1 at Ser616 and Ser637 has been extensively studied. Cyclin B-dependent kinase (CDK1) phosphorylates Drp1 at Ser616 to target Drp1 to the mitochondria and stimulates mitochondrial fission ensuring inheritance of mitochondria by daughter cells during mitosis [94, 95]. In this process another mitotic kinase Aurora A, the small Ras-like GTPase RALA, its effector RALBP1 and Mff, but not Fis1, appear to be involved [94, 96]. As the adult human heart has low mitotic potential, this regulatory event occurs infrequently in this organ. Oxidative stress induces protein kinase Cδ[delta]-mediated phosphorylation of human Drp1 at Ser616 resulting in aberrant mitochondrial fission associated with hypertension-induced brain damage [97].
Both CDK1 and cAMP-dependent protein kinase A (PKA) phosphorylate human Drp1 at Ser637 near the GED domain interfering with the interaction between GED and GTPase domains. This results in the inhibition of Drp1 GTPase activity and its recruitment to mitochondria and eventually in attenuation of mitochondrial fission [98, 99]. The PKA-mediated Drp1 phosphorylation protects mitochondria from autophagosomal degradation and enhances cell viability during nutrient starvation [100, 101]. Conversely, the phosphatase calcineurin dephosphorylates Drp1-Ser637 in a Ca2+-dependent manner targeting Drp1 to mitochondria and inducing mitochondrial fission [99, 102]. Moreover, the dephosphorylation of Drp1-Ser637 is involved in apoptotic and programmed necrotic death pathways [98, 99, 103, 104].
Drp1 is also subject of O-linked-N-acetyl-glucosamine glycosylation (O-GlcNAcylation) and S-nitrosylation [105, 106]. In cardiomyocytes, the O-GlcNAcylation of Drp1 at Thr585 and Thr586 has led to Drp1-Ser637 dephosphorylation associated with mitochondrial fragmentation and loss of membrane potential [106, 107]. The O-GlcNAcylation of OPA1 linked to mitochondrial fragmentation has also been demonstrated in neonatal cardiomyocytes [106].
Intriguingly, the phosphorylation of the same residue by calcium/calmodulin-dependent protein kinase 1α (CaMKIα[alpha]) and the Rho-associated coiled-coil-containing protein kinase1 (ROCK1) induces Drp1 recruitment to mitochondria and enhances mitochondrial fission [108, 109]. Although the reason of this seemingly different consequence of Drp1-Ser637 phosphorylation is yet unclear, it might be linked to tissue-specific response and/or phosphorylation of additional factors, which may be involved in Drp1-promoted fission [17].
Mitochondrial dynamics, mitophagy and cell death in heart failure
In contrast to other cell types and neonatal cardiomyocytes, in adult human cardiomyocytes, mitochondrial fusion and fission are very rare events due to specific intracellular mitochondrial arrangements [110]. Growing evidence suggests that the cardiomyocyte mitochondrial dynamics machinery performs additional noncanonical functions governing Ca2+ handling, mitophagy, mitochondrial quality control and cell death pathways. Nevertheless, recent conditional cardiac-specific ablation of both Mfn1 and Mfn2 has provided clear evidence that mitochondrial fusion occurs in adult mammalian cardiomyocytes, albeit at extremely slow rate [29]. Moreover, perinatal cardiac-specific deletion of both mitofusins caused early lethality due to severe mitochondrial abnormalities associated with cardiomyopathy [30]. The conditional Mfn1/Mfn2 double knockout in the adult mouse heart has led to mitochondrial fragmentation associated with severe cardiomyocyte respiratory defects culminating in HF within 6–8 weeks [29]. These findings highlight the critical role that mitofusins play in cardiac development and homeostasis.
Importantly, single knockout of Mfn1 and Mfn2 in mice has displayed distinct phenotypes. Mfn1 deficiency resulted in fragmented mitochondria and elevated apoptosis in neonatal rat cardiac myocytes, and these defects can be rescued by Mfn1 overexpression [111]. However, although mature mouse Mfn1 −/− cardiomyocytes have also accumulated fragmented mitochondria, they have paradoxically been more resistant to stress-induced mitochondrial permeability transition pore (MPTP) opening and apoptosis [112].
Unlike Mfn1 deficiency, ablation of Mfn2 in mouse heart has not impaired mitochondrial fusion as evidenced by increased mitochondrial size. However, Mfn2 −/− cardiac mitochondria displayed dissipation of mitochondrial membrane potential and elevated ROS production [113, 114]. Importantly, Mfn2-deficient mice developed cardiac hypertrophy and ventricular dysfunction with age [91]. Furthermore, cardiac-specific deletion of Mfn2, but not Mfn1, has impaired tethering of mitochondria to SR and disrupted Ca2+ handling that is critical for cardiac function [113, 114].
Another fusion protein OPA1 also plays an important role in heart physiology. OPA1 deficiency caused mitochondrial fragmentation and abnormal cristae remodeling [115]. Heterozygous OPA1 +/− mice exhibited mitochondrial dysfunction, mtDNA instability and elevated ROS production and developed cardiomyopathy [116, 117]. Consistently, cardiomyocytes derived from these animals have characterized abnormal Ca2+ handling, contractility and high susceptibility to ischemia reperfusion injury (IRI) [116]. Importantly, reduced OPA1 levels associated with accumulation of fragmented mitochondria have been reported in human failing hearts [118].
Mitochondrial fusion has traditionally been envisioned as a prosurvival antiapoptotic mechanism. Indeed, in some cell types silencing of Mfn1 or Mfn2 has enhanced cellular susceptibility to apoptotic stimuli, whereas overexpression of Mfn2 or OPA1 has attenuated apoptosis [115, 119, 120]. However, recent studies on mature cardiomyocytes unexpected effects of inhibiting mitochondrial fusion have been reported (see also above). Ablation of both mitofusins in the adult heart has led to no significant change in MPTP sensitivity, a key event in the initiation of apoptosis [29]. Similarly, although cardiac mitochondria of heterozygous Opa1+/− mice displayed disorganized mitochondrial cristae, unexpectedly they exhibited higher Ca2+ retention capacity and delayed MPTP opening under Ca2+ stimulation [117]. In addition, it has been demonstrated that mitochondrial fusion can be harmful to the cell when damaged mitochondria are fused with functional organelles due to attenuated mitophagy [121].
Prolonged and/or high-level stress can eventually lead to mitochondrial damage and dysfunction. Mitophagy induced by cardiac stress removes damaged dysfunctional mitochondria preventing thereby oxidative damage, which can otherwise initiate apoptosis and ultimately HF [122, 123]. Increasing evidence suggests that Mfn2 plays a complex role in cardiac physiology and pathophysiology orchestrating mitochondrial fusion, mitochondrial-SR Ca2+ signaling, mitochondrial quality control and cell death [28, 124, 125]. Its localization in the OMM and the ER/SR may facilitate organelles tethering and autophagosome formation and maturation during mitophagy [126]. It has also been suggested that regulation of mitophagy rather than mitochondrial remodeling per se is a primary role of Mfn2 in the adult human heart [21].
Mitofusins on the damaged mitochondria are rapidly ubiquitinated by the PINK1/Parkin complex (critical mediator of mitophagy), degraded by the proteosome, preventing fusion of dysfunctional mitochondria with the healthy mitochondrial network [127, 128]. Stabilization and accumulation of mitochondrial kinase PINK1 in damaged mitochondria is the initiating signal for translocation of the cytosolic Parkin E3 ubiquitin ligase to damaged organelles [129, 130]. Although crosstalk between PINK1 and Parkin is yet poorly understood, PINK1-mediated phosphorylation of Mfn2 is essential for Parkin recruitment to damaged mitochondria [91]. Parkin promotes ubiquitination of multiple OMM proteins in dysfunctional mitochondria, including both Mfn 1 and 2, attracting autophagosomes and initiating thereby mitophagy. It is well established that loss-of-function mutations in the PINK1 and Parkin genes cause early-onset autosomal recessive Parkinson’s disease [131, 132]. Recently, it has been shown that impairment in PINK1/Parkin-promoted mitophagy has also led to cardiac dysfunction. Indeed, Pink1 −/− mice exhibited abnormal cardiac mitochondrial function and elevated oxidative stress [133], whereas deletion of Parkin resulted in accumulation of abnormal mitochondria associated with heart damaged with age [122, 123]. Importantly, Mfn2 −/− mice displayed reduced PINK1/Parkin-mediated mitophagy associated with severe cardiac dysfunction leading to HF by 30 weeks of age [134].
Reduced OPA1 levels in the IMM of depolarized mitochondria have also contributed to the prevention of the damaged mitochondria to be fused, targeting them to mitophagy [135]. Consistently, inhibition of the autophagic processes has led to accumulation of dysfunctional mitochondria in various tissues, especially those with elevated energy demands, such as brain, heart, kidney, liver and pancreatic β [beta] cells [135–138].
Mitochondrial fission leading to organelle fragmentation is a prerequisite for mitophagy. Drp1 recruitment and Fis1 recruitment to mitochondria are the early events of the process in various cells, including cardiomyocytes [135, 139–142]. Cardiac-specific Drp1 −/− mice exhibited accumulation of dysfunctional mitochondria due to suppressed mitophagy, developed left ventricular dysfunction and died within 13 weeks. Furthermore, cardiac-specific heterozygous Drp1 +/− mice exhibit significantly greater infarct size after ischemia/reperfusion than control animals [143]. Other proteins implicated in mitochondrial dynamics are also active players in these processes forming the complex mitochondrial dynamics-mitophagy-cell death interactome [21, 144, 145].
Apoptotic stimuli have triggered mitochondrial hyperfusion followed by mitochondrial fragmentation concomitantly with OMM permeabilization and cytochrome c release [146, 147]. Consistently, fission protein Drp1 is implicated in this process and its suppression has not only resulted in reduced mitochondrial fission but also prevented cytochrome c release and subsequent apoptosis [148–151]. Of note, Drp1 depletion has not completely attenuated mitochondrial fission suggesting that additional factors contribute to this process during apoptosis [152, 153]. Drp1 collaborates with the proapoptotic Bcl-2 family proteins Bax and Bak by enhancing Bax oligomerization during apoptosis [154–156]. Apoptotic Bax activation induces Bax/Bak-mediated sumoylation of Drp1 leading to Drp1 translocation from the cytosol to mitochondria to promote mitochondrial fission [157].
Another mechanism contributing to ischemia-induced mitochondrial fragmentation has recently been suggested. It has been shown that myocardial ischemia has downregulated miR-499 leading to the activation of calcineurin [158]. Activated calcineurin dephosphorylates and activates Drp1 stimulating its recruitment to mitochondria, to promote mitochondrial fission. Usually, mdivi-1, a pharmacological Drp1 inhibitor, has prevented mitochondrial depolarization, fragmentation and ischemia-induced cell death in both HL-1 cells and adult cardiomyocytes [151, 159]. Basically, the mdivi-1 inhibition of Drp1-mediated fission has a cardioprotective effect reducing significantly myocardial infarction size after IRI [159]. Attenuation of other fission proteins, such as Fis1, Mff or MTP18, has also led to a delay in cytochrome c release and reduced apoptosis [74, 149, 160].
In addition to Bax/Bak proteins, two other members of Bcl-2 family, the BH3-only proteins Bnip3 and Nix (also known as Bnip3L), which are involved in post-infarction cardiac remodeling and cardiomyocyte death, also contribute to this complex process [21]. Nix −/− mice have developed cardiac dysfunction and hypertrophy with age, while double Nix/Bnip3 knockout mice accumulated dysfunctional mitochondria and developed cardiac dysfunction at about twice the rate than Nix −/− mice [161].
Consistent with their role in mitochondrial turnover, overexpression of Bnip3 or Nix has led to activation of PINK1/Parkin-mediated mitophagy playing a protective role [142, 162]; however, upon cardiac stress these proteins can exert detrimental effects. Bnip3 mediates cardiomyocyte death in ischemia-induced HF [163, 164], whereas Nix is upregulated in hypertrophic hearts and promotes the transition from cardiac hypertrophy to HF [165–167]. Similar to Mfn2, these proteins have dual subcellular localization to mitochondria and adjacent ER/SR. Interestingly, Nix and Bnip3 localization to the mitochondria or ER/SR determines whether they mediate cardiomyocyte death, predominantly through apoptosis or necrosis, respectively [167–169].
In summary, under physiological conditions, basal levels of mitophagy are critical for maintaining the appropriate number of functional mitochondria, preserving therefore cardiac integrity and contractile function. Mitophagy also plays an essential role in the heart adaptation to mild stress. However, upon prolonged and/or high stress, mitophagy can be detrimental to the heart. Imbalanced activation or inhibition of this process can lead to excessively reduced number of functional mitochondria or accumulation of damaged organelles, respectively, resulting in cardiac dysfunction and cardiomyocyte death (via apoptosis or necrosis) and culminating in HF (Fig. 3) [170].

Mitophagy and mitochondrial quality control. a Normal mitophagy begins with the initiation and elongation of a double-membraned autophagic vesicle. The vesicle then sequesters and engulfs mitochondria for degradation. Proper regulation of mitophagy leads to mitochondrial quality control and cellular homeostasis. b Increased mitophagy may greatly reduce the pool of functional mitochondria. With too few mitochondria, the cell loses its ability to produce sufficient energy and eventually dies. c Reduction in mitophagy causes accumulation of dysfunctional mitochondria. The dysfunctional mitochondria generate excessive ROS and release prodeath proteins, triggering rapid cell death. From Shires and Gustafsson [170] with permission of Springer Publishing Co.
Discussion
Over the past decade, cardiac mitochondria have emerged as critical integrators of energy production, ROS generation, Ca2+ handling and multiple signaling and cell death pathways. Tightly balanced processes of mitochondrial fusion and fission contribute to the multifaceted role that mitochondria play in myocardial physiology. Great progress has been recently achieved deciphering the complex multiprotein machineries that promote mitochondrial dynamics, and growing evidence suggests that defects in the core components of these machineries can cause alteration in mitochondrial structure and function leading eventually to various human disorders, including HF.
The most established causative link between mutations in the genes encoding proteins, which mediate mitochondrial fusion and fission, and pathological conditions has been demonstrated in inherited neurological and neurodegenerative disorders, including autosomal dominant optic atrophy, Charcot–Marie–Tooth neuropathy and Wolf–Hirschhorn syndrome. Abnormalities in mitochondrial dynamics have also been associated with age-related progressive neurodegenerative disorders, such as Alzheimer’s, Parkinson’s and Huntington’s diseases. Notably, patients with Parkinson’s and Danon’s diseases, which are characterized by impaired mitophagy, develop cardiomyopathy and HF [171, 172].
Although the human heart has a great density of mitochondria and is characterized by high levels of the major proteins implicated in mitochondrial dynamics, we have only begun to uncover the multifaceted role of the fusion–fission processes in cardiac physiology and pathophysiology. The simplified view that upregulation of the fusion machinery is cardioprotective, while upregulation of the fission factors inevitably lead to mitochondrial fragmentation and trigger cell death, has recently been challenged. Similarly, mitophagy, a highly complex and tightly regulated pathway, is mediated by the coordinated action of multiple proteins. As a mitochondrial quality control mechanism mitophagy plays a critical cardioprotective role by removing dysfunctional mitochondria, although when impaired it can be detrimental to the heart.
The role that fusion protein Mfn2 and other mitochondrial dynamics factors play in mitophagy and stress-induced cardiomyocyte death remains controversial and requires further investigation. Recent evidence suggests that in the human heart Mfn2 interacts with various proteins and primarily functions as a key orchestrator of mitochondrial fate and cardiac homeostasis [21, 125]. However, the precise molecular mechanisms underlying the interaction of mitochondrial dynamics proteins with mitophagy and cell death factors, and their involvement in the development and progression of HF remain to be determined.
Pharmacological targeting of components of the mitochondrial fusion and fission machineries that shift the balance toward normal mitochondrial numbers, morphology and function may be a novel therapeutic modality for CVD, including HF. One of the first evidence that a specific Drp1 inhibitor, mdivi-1, with direct effects on mitochondrial fission can act as a preconditioning agent, protecting the myocardium against IRI, is very promising [159]. Another recent example of beneficial targeting of mitochondrial dynamics is the generation of transgenic mice overexpressing miR-499, which exhibited protection against post-ischemic cardiomyocyte death, myocardial infarction and ventricular remodeling [158]. The emerging critical role of Mfn2 and OPA1 in the differentiation of embryonic stem cells into cardiomyocytes may also be important for the development of innovative cell-based therapy for HF [173]. Lastly, further research is needed to establish whether the targeting of mitochondrial fusion–fission and mitophagy machineries can restore the number, morphology and function of this critical organelle and whether it could be translated into clinically relevant therapy for HF.
Conclusions
-
Mitochondria are able to vary their morphology through complex processes of fusion and fission. These processes also allow the transmission of signals and the exchange of metabolites within the cell.
-
Mitochondrial fusion and fission are implicated in numerous biological processes including embryonic development and cell death.
-
It is important to understand at which stage mitophagy is adaptive and when it is maladaptive, since excessive mitophagy may deplete the mitochondrial pool, which if falling below required level for cardiac contractile activity or maintenance of cellular integrity will lead to cardiac dysfunction and to the death of individual cardiomyocytes.
-
Changes in mitochondrial morphology may contribute to cardiac development, the myocardial response to IRI, and HF.
-
Failure to remove damaged mitochondria might increase cellular death from excessive ROS generated by defective mitochondria.
-
Targeting the mitochondrial fusion–fission and mitophagy machineries may restore the number, morphology and function of this organelle. However, further research is needed to translate these findings into successful therapy for HF.
References
Hoppel CL, Tandler B, Fujioka H, Riva A (2009) Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol 41:1949–1956
Ong SB, Hausenloy DJ (2010) Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88:16–29
Ventura-Clapier R, Garnier A, Veksler V, Joubert F (2011) Bioenergetics of the failing heart. Biochim Biophys Acta 1813:1360–1372
Soubannier V, McBride HM (2009) Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta 1793:154–170
Hausenloy DJ, Ruiz-Meana M (2010) Not just the powerhouse of the cell: emerging roles for mitochondria in the heart. Cardiovasc Res 88:5–6
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148:1145–1159
Brown DA, O’Rourke B (2010) Cardiac mitochondria and arrhythmias. Cardiovasc Res 88:241–249
Cadenas S, Aragones J, Landazuri MO (2010) Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res 88:219–228
Rosca MG, Hoppel CL (2010) Mitochondria in heart failure. Cardiovasc Res 88:40–50
Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B et al (2012) Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep 14:532–539
Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89:799–845
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065
Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505:335–343
Archer SL (2013) Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369:2236–2251
Hoppins S (2014) The regulation of mitochondrial dynamics. Curr Opin Cell Biol 29:46–52
Elgass K, Pakay J, Ryan MT, Palmer CS (2013) Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 1833:150–161
Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A et al (2007) Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med 4(Suppl 1):S60–S67
Youle RJ, Karbowski M (2005) Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 6:657–663
Hall AR, Burke N, Dongworth RK, Hausenloy DJ (2014) Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol 171:1890–1906
Dorn GW 2nd, Kitsis RN (2015) The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res 116:167–182
Itoh K, Nakamura K, Iijima M, Sesaki H (2013) Mitochondrial dynamics in neurodegeneration. Trends Cell Biol 23:64–71
Ong SB, Hall AR, Hausenloy DJ (2013) Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal 19:400–414
Dorn GW 2nd (2015) Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med 7:865–877
Sharp WW, Archer SL (2015) Mitochondrial dynamics in cardiovascular disease: fission and fusion foretell form and function. J Mol Med (Berl) 93:225–228
Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS (2000) Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32:97–104
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Dorn GW 2nd, Maack C (2013) SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol 55:42–49
Chen Y, Liu Y, Dorn GW 2nd (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109:1327–1331
Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I et al (2012) Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111:1012–1026
Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P et al (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33:2798–2813
Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A et al (2015) Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol 35:211–223
Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21:273–285
Westermann B (2008) Molecular machinery of mitochondrial fusion and fission. J Biol Chem 283:13501–13505
Schmid SL, Frolov VA (2011) Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol 27:79–105
Ishihara N, Otera H, Oka T, Mihara K (2013) Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal 19:389–399
Ranieri M, Brajkovic S, Riboldi G, Ronchi D, Rizzo F et al (2013) Mitochondrial fusion proteins and human diseases. Neurol Res Int 2013:293893
Rojo M, Legros F, Chateau D, Lombes A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115:1663–1674
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE et al (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM et al (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305:858–862
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C et al (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26:207–210
Olichon A, Baricault L, Gas N, Guillou E, Valette A et al (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278:7743–7746
Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B et al (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 109:584–591
Pellegrini L, Passer BJ, Canelles M, Lefterov I, Ganjei JK et al (2001) PAMP and PARL, two novel putative metalloproteases interacting with the COOH-terminus of Presenilin-1 and -2. J Alzheimers Dis 3:181–190
Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L et al (2006) Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126:163–175
Ishihara N, Fujita Y, Oka T, Mihara K (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25:2966–2977
Griparic L, Kanazawa T, van der Bliek AM (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol 178:757–764
Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S et al (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187:1023–1036
Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187:959–966
Song Z, Chen H, Fiket M, Alexander C, Chan DC (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178:749–755
Baker MJ, Tatsuta T, Langer T (2011) Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol doi:10.1101/cshperspect.a007559
Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A et al (2006) Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281:37972–37979
Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20:3525–3532
Sesaki H, Jensen RE (2001) UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol 152:1123–1134
Sesaki H, Jensen RE (2004) Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem 279:28298–28303
Guillery O, Malka F, Landes T, Guillou E, Blackstone C et al (2008) Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100:315–325
Otera H, Ishihara N, Mihara K (2013) New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833:1256–1268
Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143:351–358
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4:815–826
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Bhar D, Karren MA, Babst M, Shaw JM (2006) Dimeric Dnm1-G385D interacts with Mdv1 on mitochondria and can be stimulated to assemble into fission complexes containing Mdv1 and Fis1. J Biol Chem 281:17312–17320
Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE et al (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem 285:32494–32503
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S et al (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE et al (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12:565–573
Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667
Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151:367–380
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379
Suzuki M, Jeong SY, Karbowski M, Youle RJ, Tjandra N (2003) The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol 334:445–458
Suzuki M, Neutzner A, Tjandra N, Youle RJ (2005) Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J Biol Chem 280:21444–21452
Zhang Y, Chan DC (2007) Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci USA 104:18526–18530
Jofuku A, Ishihara N, Mihara K (2005) Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem Biophys Res Commun 333:650–659
Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19:2402–2412
Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M et al (2010) A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet 6:e1001000
Loson OC, Liu R, Rome ME, Meng S, Kaiser JT et al (2014) The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 22:367–377
Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K et al (2014) Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol 204:477–486
Zorzano A (2009) Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab 34:433–439
Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E et al (2013) PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 187:865–878
Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL (2015) Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl) 93:229–242
Garedew A, Andreassi C, Moncada S (2012) Mitochondrial dynamics, biogenesis, and function are coordinated with the cell cycle by APC/C CDH1. Cell Metab 15:466–479
Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M et al (2012) Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell 47:547–557
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ et al (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 105:1638–1643
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/Parkin pathway. PLoS ONE 5:e10054
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH et al (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/Parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861–4870
Glauser L, Sonnay S, Stafa K, Moore DJ (2011) Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118:636–645
Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP et al (2011) Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE 6:e16746
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107:378–383
Chen Y, Dorn GW 2nd (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340:471–475
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC et al (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ et al (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282:11521–11529
Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X et al (2012) Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110:1484–1497
Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD et al (2011) RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 13:1108–1115
Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D (2011) Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol Biol Cell 22:256–265
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282:21583–21587
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8:939–944
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598
Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108:10190–10195
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C et al (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105:15803–15808
Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G et al (2010) Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med 2:490–503
Wang Z, Jiang H, Chen S, Du F, Wang X (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148:228–243
Chang CR, Blackstone C (2010) Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci 1201:34–39
Makino A, Suarez J, Gawlowski T, Han W, Wang H et al (2011) Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol 300:R1296–R1302
Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H et al (2012) Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287:30024–30034
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y et al (2008) CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182:573–585
Wang W, Wang Y, Long J, Wang J, Haudek SB et al (2012) Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15:186–200
Westermann B (2002) Merging mitochondria matters: cellular role and molecular machinery of mitochondrial fusion. EMBO Rep 3:527–531
Li J, Zhou J, Li Y, Qin D, Li P (2010) Mitochondrial fission controls DNA fragmentation by regulating endonuclease G. Free Radic Biol Med 49:622–631
Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF Jr et al (2012) Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 302:H167–H179
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I et al (2011) Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol 31:1309–1328
Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N et al (2012) Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res 111:863–875
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV et al (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189
Chen L, Liu T, Tran A, Lu X, Tomilov AA et al (2012) OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc 1:e003012
Piquereau J, Caffin F, Novotova M, Prola A, Garnier A et al (2012) Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 94:408–417
Chen L, Gong Q, Stice JP, Knowlton AA (2009) Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84:91–99
Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279:52726–52734
Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H (2005) Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 280:25060–25070
Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd (2014) Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 114:257–265
Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E et al (2013) Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun 4:2308
Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN et al (2013) Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288:915–926
Dorn GW 2nd (2013) Mitochondrial dynamics in heart disease. Biochim Biophys Acta 1833:233–241
Dorn GW 2nd (2013) Mitochondrial dynamism and cardiac fate—a personal perspective. Circ J 77:1370–1379
Zhao T, Huang X, Han L, Wang X, Cheng H et al (2012) Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem 287:23615–23625
Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA 107:5018–5023
Ziviani E, Whitworth AJ (2010) How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy 128:660–662
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Eiyama A, Okamoto K (2015) PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 33:95–101
Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev 91:1161–1218
Deas E, Wood NW, Plun-Favreau H (2011) Mitophagy and Parkinson’s disease: the PINK1-Parkin link. Biochim Biophys Acta 1813:623–633
Billia F, Hauck L, Konecny F, Rao V, Shen J et al (2011) PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108:9572–9577
Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS et al (2014) Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115:348–353
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T et al (2010) Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6:600–606
Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I et al (2011) Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22:902–913
Jung HS, Lee MS (2009) Macroautophagy in homeostasis of pancreatic beta-cell. Autophagy 5:241–243
Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP et al (2005) Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol 15:2112–2118
Gomes LC, Scorrano L (2008) High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta 1777:860–866
Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI et al (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3:e3257
Lee Y, Lee HY, Hanna RA, Gustafsson AB (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301:H1924–H1931
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S et al (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–278
Tolkovsky AM (2009) Mitophagy. Biochim Biophys Acta 1793:1508–1515
Gomes LC, Scorrano L (2013) Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta 1833:205–212
Martinou JC, Youle RJ (2006) Which came first, the cytochrome c release or the mitochondrial fission? Cell Death Differ 13:1291–1295
Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y et al (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28:1589–1600
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG et al (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1:515–525
Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15:5001–5011
Germain M, Mathai JP, McBride HM, Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J 24:1546–1556
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C et al (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO et al (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11:958–966
Landes T, Martinou JC (2011) Mitochondrial outer membrane permeabilization during apoptosis: the role of mitochondrial fission. Biochim Biophys Acta 1813:540–545
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22:1577–1590
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S et al (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142:889–901
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21:92–101
Wasiak S, Zunino R, McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177:439–450
Wang JX, Jiao JQ, Li Q, Long B, Wang K et al (2011) miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 17:71–78
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM et al (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022
Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J et al (2005) The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci 118:3049–3059
Dorn GW 2nd (2010) Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3:374–383
Ding WX, Ni HM, Li M, Liao Y, Chen X et al (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285:27879–27890
Diwan A, Krenz M, Syed FM, Wansapura J, Ren X et al (2007) Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117:2825–2833
Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM et al (2006) Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem 281:1442–1448
Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G et al (2002) Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8:725–730
Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA et al (2004) Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res 95:1200–1206
Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN et al (2008) Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 117:396–404
Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A et al (2009) Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 119:203–212
Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ et al (2010) Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA 107:9035–9042
Shires SE, Gustafsson AB (2015) Mitophagy and heart failure. J Mol Med (Berl) 93:253–262
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S et al (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406:906–910
Zesiewicz TA, Strom JA, Borenstein AR, Hauser RA, Cimino CR et al (2004) Heart failure in Parkinson’s disease: analysis of the United States medicare current beneficiary survey. Parkinsonism Relat Disord 10:417–420
Kasahara A, Cipolat S, Chen Y, Dorn GW 2nd, Scorrano L (2013) Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and notch signaling. Science 342:734–737
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Marín-García, J., Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail Rev 21, 123–136 (2016). https://doi.org/10.1007/s10741-016-9530-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-016-9530-2