
Abstract
The process of muscle remodeling lies at the core of most cardiovascular diseases. Cardiac adaptation to pressure or volume overload is associated with a complex molecular change in cardiomyocytes which leads to anatomic remodeling of the heart muscle. Although adaptive at its beginnings, the sustained cardiac hypertrophic remodeling almost unavoidably ends in progressive muscle dysfunction, heart failure and ultimately death. One of the features of cardiac remodeling is a progressive impairment in mitochondrial function. The heart has the highest oxygen uptake in the human body and accordingly it has a large number of mitochondria, which form a complex network under constant remodeling in order to sustain the high metabolic rate of cardiac cells and serve as Ca2+ buffers acting together with the endoplasmic reticulum (ER). However, this high dependence on mitochondrial metabolism has its costs: when oxygen supply is threatened, high leak of electrons from the electron transport chain leads to oxidative stress and mitochondrial failure. These three aspects of mitochondrial function (Reactive oxygen species signaling, Ca2+ handling and mitochondrial dynamics) are critical for normal muscle homeostasis. In this article, we will review the latest evidence linking mitochondrial morphology and function with the process of myocardial remodeling and cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease is a growing health concern worldwide. Heart failure (HF), the common final stage of most cardiovascular diseases, shows a steadily growing prevalence and constitutes a leading cause of death in most of the occidental countries [1]. A core process linking cardiovascular diseases such as hypertension, coronary artery disease and valvular disease to HF is the abnormal remodeling of heart muscle in response to different noxas. Although initially adaptive, cardiomyocyte response to pressure or volume overload is associated with deep molecular changes leading to fetal gene expression, impaired contractile function, abnormal vascularization, altered extracellular matrix composition, fibrosis and profound metabolic abnormalities which almost unavoidably end in progressive HF [2, 3].
Because of the significant economic burden associated with HF, interventions to stop or revert pathological remodeling appear as attractive targets to delay disease progression. Current therapy in HF is aimed to avoid cardiac remodeling via neurohumoral blockade; however, despite the proven beneficial effects of drugs acting on the renin-angiotensin-aldosterone system (i.e. angiotensin converting enzyme inhibitors, AT1 receptor antagonists or aldosterone receptor antagonists) or in the adrenergic system (i.e. beta-adrenergic receptor blockers), mortality and morbidity rates associated with HF remain high, thus showing the need for novel approaches to treat the disease. An emerging therapeutic target is the metabolic derangement, which characterizes HF. In fact, cardiac tissue depends largely on mitochondrial metabolism in order to sustain its high energetic demands. The failing cardiomyocyte has been characterized as an “engine out of fuel” [3]. This energetic impairment is associated with abnormal mitochondrial dynamics, increased oxidative stress and abnormal Ca2+ handling [4] which seems to play a critical role on the myocardial remodeling process leading to HF. In this article we will review the latest evidence linking abnormal mitochondrial morphology and function with myocardial remodeling, emphasizing new emerging strategies to avoid HF development and progression.
Mitochondria, Oxidative Stress, and Muscle Remodeling
Reactive oxygen species (ROS) have a dual role in cell signaling. A controlled production of ROS acts as a secondary messenger amplifying signals that are crucial for normal cell function. In contrast, an overt imbalance between ROS production and the cell antioxidant defense systems leads to a breakage from normal homeostasis (oxidative stress) closely related to a wide range of cardiac diseases.
Within the cardiovascular system there are two major sources of ROS: cytosolic enzymes, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase, and mitochondria. While cytoplasmic ROS are usually associated with signaling, mitochondrial ROS are primarily a byproduct of mitochondria’s role as the major site of oxygen consumption in the cell. During cardiac remodeling and heart failure, both cytosolic and mitochondrial ROS becomes deregulated leading to oxidative damage, mitochondrial dysfunction and ultimately cell death.
It is well established that ROS play a role in myocardium remodeling and progression to heart failure [5], although their effect likely varies depending on ROS type and concentration. Under physiological conditions, mitochondria are responsible for almost all superoxide production in cardiomyocytes; however, during hypertrophy, cytosolic sources such as NADPH oxidase also contribute to increased oxidative stress [6]. In a cardiomyocyte model, Kwon et al. demonstrated that different concentrations of ROS could differentially activate pathways to induce hypertrophy or apoptosis: low ROS concentrations promoted increased survival and hypertrophy through extracellular signal-regulated kinase 1/2 (ERK1/2) activation, while higher concentrations activated stress kinases like c-Jun N-terminal kinase (JNK) and p38-mitogen activated protein kinase (MAPK) [7]. Activation of JNK may link the hypertrophy and mitochondrial dysfunction seen in heart failure; in fact, JNK activation promotes not only cardiomyocyte hypertrophy but also the activation of autophagy through Bcl-2 and nineteen-KDa interacting protein-3 (BNIP3), which ultimately leads to apoptosis and mitochondrial selective autophagy (mitophagy) [8•, 9]. In an intriguing paper, Vacek et al. showed that increased mitophagy may lead to metalloproteinase activation, a key element in cardiovascular remodeling as extracellular matrix degradation leads to increased collagen deposit and impaired cardiomyocyte coupling [10].
Interestingly, the activation of ROS-dependent pathways seems to be characteristic of pathological hypertrophy [11]. Physiological hypertrophy, as seen in high-trained athletes or pregnancy, appears to depend on different signaling pathways such as insulin-like growth factor-1 (IGF-1)/protein kinase Akt and the induction of peroxisome proliferator activator γ coactivator 1α (PGC-1α) leading to increased mitochondrial biogenesis. In contrast, there is a marked reduction in PGC-1α/estrogen-related receptor α (EERα) seen in pathological hypertrophy [12].
Mitochondria have developed specific strategies to avoid ROS induced damage. One of the most interesting is associated with deacetylase Sirtuin 3 (Sirt3), which resides in the mitochondrial membrane and is closely related to redox environment regulation. Sirt3 modifies ROS production both directly, regulating mitochondrial superoxide dismutase (SOD) and isocitrate dehydrogenase (IDH), and indirectly, regulating mitochondrial electron transport chain (mETC) activity, lipid processing and inhibiting mitochondrial permeability transition pore (MPTP) opening [13]. In fact, cardiac hypertrophy enhances Sirt3 expression. Furthermore, Sirt3 knock-out animals subjected to the same stimuli displayed an exacerbated hypertrophic response, suggesting that augmented Sirt3 levels are a compensatory response [14]. Another protein likely involved in maintaining mitochondrial function in the face of increased oxidative stress is the PTEN-induced kinase (PINK). This protein, usually associated with mitophagy, is markedly decreased in advanced stages of heart failure. PINK knock-out mice display spontaneous cardiac hypertrophy, increased cardiomyocyte oxidative stress, impaired mitochondrial function and increased myocyte apoptosis [15].
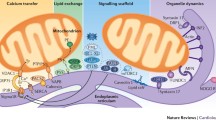
The relationship between cardiac remodeling and hypertrophy is reciprocal. Mitochondria are not only the main source of ROS in the cardiomyocyte, they are also one of the main targets of oxidative damage. Mitochondrial DNA (mtDNA) is particularly prone to oxidative damage due to its lack of histones and the high levels of ROS generation in the organelle matrix [16]. This oxidative damage may also affect critical steps within the Krebs cycle and also mtDNA polymerase γ, slowing mtDNA replication and eventually leading to inhibition of oxidative phosphorylation [17]. As mtDNA encodes proteins involved in complexes I and III of the mETC, a vicious circle is established leading to progressive mitochondrial dysfunction, as seen in advanced stages of heart failure [18]. The importance of mtDNA has been further supported by the recent discovery that impaired autophagy of mitochondrial DNA released from damaged mitochondria may play a role in the development of the inflammatory response that accompanies certain forms of myocarditis and heart failure [19]. The participation of mitochondrial ROS in cardiac remodeling is summarized in Fig. 1a.

a. Oxidative stress and cardiac remodeling. An increase in mitochondrial reactive oxygen species (ROS) promotes the activation of the mitogen-activated protein kinases ERK, p38 and JNK inducing cardiac hypertrophy. It has recently shown that JNK can activate mitophagy, giving another pathway to promote cardiac failure through BNIP3. Other internal mitochondrial proteins can also regulate these pathways; both PINK and Sirt3 have a protective role against hypertrophy. Moreover the accumulation of ROS inside mitochondria induces mtDNA damage and impairs mitochondrial electron transport chain, increasing ROS production which ultimately leads to a vicious circle inducing further damage. Recently it has been demonstrated that mtDNA itself released from damaged organelles may be responsible of myocardial damage by activation of the innate immune response. b. Ca2+ signaling and cardiac remodeling. Cytoplasmic Ca2+ rise activates the Ca2+ -dependent proteins CAMKII, calcineurin (CaN) and protein kinase C (PKC), which in turn induces a characteristic genetic program involved on cardiac hypertrophy development. Also, the activation of CAMKII and CaN promotes mitochondrial fission and MPTP opening; the release of cytochrome c activates cardiomyocyte apoptosis, further contributing to the remodeling process. c. Mitochondrial dynamics and cardiac remodeling. The participation of mitochondrial dynamics on remodeling seems to depend on the nature of the injury. In pressure overload models, a decrease in Mfn2 or Opa1 promotes cardiac hypertrophy. Paradoxically, Mfn2 knock down improves the functional recovery after myocardial ischemia/reperfusion. A decrease in mitochondrial fission - via a dominant negative form of Drp1 or pharmacological inhibitors such as Mdivi - decrease cardiac remodeling irrespective of the mechanism (pressure overload or ischemia/reperfusion)
Considering the involvement of mitochondrial ROS in cardiac hypertrophy and remodeling, several investigators have targeted mitochondrial ROS as an approach to preventing cardiac failure. Dai et al. recently demonstrated that mitochondrial-targeted catalase partially protected the heart from failure induced by transverse aortic constriction and consistently attenuated mitochondrial proteome remodeling [20•]. These findings were perfectly mimicked by the mitochondrial-targeted antioxidant SS-31, but not by a non-specific antioxidant such as N-acetylcysteine, reinforcing the concept that mitochondrial ROS are causative in the development of heart failure [21].
Mitochondrial Ca2+ Handling in Muscle Remodeling and Cardiovascular Disease
The major function of the heart is to pump blood throughout the circulatory system providing oxygen and nutrients to all tissues and removing unwanted metabolites. This pumping activity requires essential beat-to-beat rhythmic oscillations in cytosolic Ca2+ of individual cardiomyocytes, that drives changes in myofilament interactions promoting contraction and cell shortening [22]. However, in the last two decades a new understanding of the role of Ca2+ in programming reactive hypertrophic signaling has emerged. Ca2+ is stored mainly in the sarcoplasmic reticulum (SR), which generates Ca2+ signals through controlled release of stored Ca2+ in to the cytoplasm. In this particular function, two Ca2+ channels stand out, the inositol 1,4,5-triphosphate receptor (IP3R) and the ryanodine receptor (RyR). Following a release from the SR to the cytoplasm, Ca2+ is swiftly pumped back by the action of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), thus maintaining low cytoplasmic concentrations and preventing the depletion of SR Ca2+ [22].
Diverse signaling proteins activated by an increase in cytosolic Ca2+ have been associated with the development of cardiac hypertrophy, such as calcineurin (CaN), calmodulin-dependent protein kinase (CAMKII) and protein kinase C (PKC). For a more comprehensive review see references [23•, 24•, 25•]. Notably, these proteins are also associated with mitochondrial-dependent cell death. Cardiomyocytes expose to isoproterenol the pro-apoptotic protein Bad dephosphorylate, which then translocates to mitochondria to promote cell death. CaN inhibitors, such as FK506 and cyclosporine, inhibit dephosphorylating of Bad and increase cardiomyocyte survival [26]. In ischemic/reperfusion injury, CAMKII-dependent phosphorylation of phospholamban triggers mitochondria-dependent cell death. The use of KN93 (a CAMKII inhibitor) reduces the release of cytochrome c from mitochondria, mitochondria swelling and lactic dehydrogenase (LDH) release from the cell [27]. On the other hand, post-conditioning of cardiomyocytes protects from cell death through a PKC-ξ interaction with a Ca2+ sensing receptor, that inhibits the SR-dependent increase in cytoplasmic Ca2+ and mitochondrial Ca2+ overload [28]. The main pathways connecting Ca2+ handling and muscle remodeling are illustrated in Fig. 1b.
These observations have fostered an interest in mitochondrial Ca2+ handling as a therapeutic target in cardiovascular disease. For example, mitochondria from diabetic hearts have an increased sensitivity to MPTP opening in response to ischemic/reperfusion injury compared to mitochondria from non-diabetic hearts. Blocking the Ca2+ influx with the mitochondrial Ca2+ uniporter blocker minocycline or reducing ROS levels with the mitochondrial-targeted peptide MTP-131 reduces infarct size in both normal and diabetic hearts [29] demonstrating the importance of mitochondrial Ca2+ in this process.
Mitochondria themselves are important targets of Ca2+ signaling. Once in the mitochondrial matrix, Ca2+ has several mechanisms through which it can regulate mitochondrial metabolism. It was shown several decades ago that Ca2+ directly activates pyruvate dehydrogenase phosphatase and three Krebs cycle dehydrogenases [30]. Ca2+ also increases activity of the F1/F0 ATPase [31] and the ATP/ADP carrier [32]. Thus, mitochondrial Ca2+ is essential for cell bioenergetics and a raise in mitochondrial Ca2+ concentration can stimulate Krebs cycle activity, increasing NADH levels leading to an increase in ATP synthesis [33•, 34].
Mitochondrial Ca2+ uptake is a key determinant for the regulation of mitochondrial metabolism by this ion. Ca2+ entrance to the mitochondria is mainly sustained by the mitochondrial Ca2+ uniporter (mCU), whose molecular identity has been recently discovered [35]. mCU is a highly selective Ca2+ channel, but has a low affinity for Ca2+ [36]. As a result, a high concentration of cytosolic Ca2+ in the proximity of mitochondria must occur to induce Ca2+ entry. To solve this, mitochondria are strategically localized in high Ca2+ concentration micro domains near the Ca2+ release channels within ER [37, 38]. The close contact formed between ER and mitochondria is regulated by a great variety of proteins located in the interface of the two organelles, including IP3R, RyR, SERCA and mitofusin-2 (Mfn-2) [39]. The evidence for this coupling in cardiomyocytes is demonstrated by the difficulty of isolating purified cardiac mitochondria free from associated SR vesicles, suggesting a direct physical coupling. These SR particles can also transfer Ca2+ to the mitochondria and activate oxidative metabolism [40]. On the other hand, the main mechanism for efflux of mitochondrial Ca2+ in cardiomyocytes is a Ca2+/Na+ exchanger, which extrudes mitochondrial Ca2+ in exchange for Na+ [41].
In the heart, where the energy demand is constant, the fine and effective regulation of mitochondrial metabolism is of utmost importance. Work done in neonatal and adult cardiomyocytes have shown that mitochondrial Ca2+ levels increase in a beat-to-beat fashion, mirroring cytoplasmic Ca2+ levels [42, 43]. Interestingly, in beating adult cardiomyocytes, cytoplasmic and mitochondrial ATP levels remain constant even after an increase in workload, suggesting a tight coupling between energy demand and production. In response to an abrupt stimulation from rest, cardiomyocytes will show a drop in mitochondrial ATP, followed by an increase over basal levels. This reduction in mitochondrial ATP levels is shadowed by an increase in mitochondrial Ca2+ [43]. Similar results have been obtained measuring changes in NADH levels in adult rat cardiomyocytes [41]. During increased workload, mitochondrial Ca2+ levels are elevated, while NADH levels remains unaltered. However, if Ca2+ entry to the mitochondria is blocked using mCU inhibitors or if Ca2+ extrusion from the mitochondria is enhanced through the mitochondrial Ca2+/Na+ exchanger by increasing cytoplasmic Na+ concentration, mitochondria cannot load Ca2+ efficiently in response to the increased workload, and NADH levels fall evidencing an energetically compromised cell [41]. Together, these results suggest that Ca2+ participates in the fine regulation of mitochondrial metabolism and ATP synthesis.
There are several reports showing that mitochondrial metabolism is altered in heart failure [44, 45]. Interestingly, cardiomyocytes from failing hearts also show a reduction in mitochondrial Ca2+ increases after electrical or adrenergic stimulation compared to the response of normal cardiomyocytes, suggesting that the reduction in mitochondrial metabolism may be due to a loss in mitochondrial Ca2+ [46]. Furthermore, in failing hearts there are reported important alterations in several Ca2+ channels and pumps controlling cytoplasmic Ca2+ handling that could explain the reduction in Ca2+ influx to the mitochondria [22]. Liu and O´Rourke showed that promoting the accumulation of mitochondrial Ca2+ via the inhibition of the Ca2+/Na+ exchanger prevented the reduction in mitochondrial metabolism in failing hearts [47]. Thus, the recovery of proper Ca2+ handling in mitochondria of failing hearts emerges as an option to prevent the detrimental effects of this condition. In this regard, IGF-1 has been shown to induce mitochondrial Ca2+ uptake and mitochondrial respiration and prevent a fall in ATP in nutritionally stressed cardiomyocytes, reducing cell death [48]. Also, Zhang et al, demonstrated a beneficial role for cardiac-specific overexpression of IGF-1 in attenuating or preventing the contractile and metabolic dysfunction induced by high-fat diet [49]. It remains to be determined if targeting Ca2+-dependent mitochondrial function is therapeutically beneficial in the clinical setting of cardiovascular diseases.
Mitochondrial Dynamics and Cardiovascular Disease
The traditional concept of mitochondria as static, isolated organelles has been challenged in recent decades as the mechanisms regulating mitochondrial function have been discovered. Nowadays, we understand mitochondria as a network under constant remodeling by the processes of mitochondrial fusion and fission. These processes are known to be essential for cell survival, as knocking out various protein components of the machinery controlling mitochondrial dynamics usually leads to a lethal phenotype [50, 51]. In fact, mitochondrial dynamics participate in processes such as mitochondrial biogenesis, mtDNA maintenance, cell metabolism, cell proliferation, cell survival, and ultimately cell death [52]. Although the regulation of mitochondrial dynamics is beyond the scope of this review, several recent reviews provide a thorough analysis of this topic [53, 54•].
Mitochondrial fusion is a two-stage process, involving outer membrane fusion (depending on the large GTPases Mfn1 and Mfn2) and inner membrane fusion, which depend on the dynamin-related protein Opa1. Mfn2 is abundantly expressed in the heart and is involved critically in cardiomyocyte apoptosis [55]. Overexpression of Mfn2 promotes mitochondrial fusion and activates the intrinsic apoptotic pathway, as seen in ischemia/reperfusion models [56]. Conditional knock-out of Mfn2 on adult cardiomyocytes seems to foster mitochondrial tolerance to Ca2+-induced MPTP opening and improves post- reperfusion recovery in an in vivo model [57]. Paradoxically, a dominant-negative form of Mfn2 increases ceramide-induced apoptosis in cardiomyocytes [58], which agrees with previous reports describing an anti-apoptotic role for proteins involved in mitochondrial fusion [59]. These conflicting results may be explained by the role of Mfn2 in multiple signaling pathways beyond mitochondrial shaping. In fact, a particularly intriguing role of Mfn2 involves establishing points of contact between mitochondria and ER. Recently, our group reported that during early stages of ER stress, mitochondria underwent perinuclear fusion, that was associated with increases in ATP levels, oxygen consumption, reductive power and mitochondrial Ca2+ uptake [60]. Loss of Mfn2 leads to decreased mitochondria-ER contacts, which may hamper the response to ER stress such as seen in cardiac hypertrophy [61]. Changes in the mitochondria-ER coupling appear also to be involved in pulmonary hypertension, a disease characterized by excessive proliferation of pulmonary artery vascular cells. Disruption of mitochondria-ER coupling by knocking out the ER-shaping protein Nogo B lead to resistance to hypoxia-induced pulmonary artery hypertension [62•]. Mounting evidence suggests a critical role of mitochondria-ER coupling in cell homeostasis [63]; however, the full implications of is interaction remain largely unknown.
Opa1 is the primary protein responsible for inner mitochondrial membrane fusion and cristae structure. During heart failure, Opa1 levels markedly decrease promoting a phenotype characterized by small mitochondria with few cristae; although the mechanisms leading to the decrease in Opa1 levels are unknown, no significant changes were seen in mRNA content, suggesting a post-translational mechanism [64•]. Interestingly, this phenotype is restricted to pathological hypertrophy/heart failure; in physiological hypertrophy, mitochondrial biogenesis is enhanced and the aforementioned changes are not seen [65].
Recently, data with haploinsufficient Opa1+/- mice have shown altered morphology of the mitochondrial network and altered MTMP function. Surprisingly, mitochondria in the heterozygous mice were larger and clustering of fused mitochondria was observed when compared with wild-type controls. Opa1+/- mice, after 6 weeks of transverse aortic constriction (TAC), had two-fold greater hypertrophic growth and a significantly reduced ejection fraction when compared with wild type mice [66]. This new evidence further reinforces the observations already described for Mfn2 that enhanced fusion of mitochondria may be deleterious in the onset of pressure overload.
Mitochondrial fission depends on the large dynamin-related GTPase Drp1. This cytosolic protein migrates to mitochondria and polymerizes forming spiral structures that constrict the organelle leading to its fission [4]. Other outer mitochondrial membrane proteins, such as mitochondrial fission factor (Mff) or mitochondria fission 1 protein (Fis1), were also required for mitochondrial fission in mammals serving as scaffolds for assembly of the fission machinery [67]. Mutations in Drp1 results in a phenotype characterized by elongated mitochondria and impaired ATP synthesis leading to dilated cardiomyopathy in mice [68]. However, hypertrophic cardiomyocytes usually present small mitochondria, with decreased levels of Mfn2 and increased Drp1 and Fis1 expression [69]. In fact, mitochondrial fission may serve as an elegant link between ROS signaling and Ca2+: increased Ca2+ promotes Drp1-dependent mitochondrial fission, increasing ROS production, as is seen in heart failure. Furthermore, the pro-hypertrophic Ca2+-dependent protein CaN induces the fragmentation of mitochondria by promoting Drp1 dephosphorylation and its translocation towards mitochondria [70]. Concordant with these observations, the expression of a dominant-negative form of Drp1 (DrpK38A) avoids ischemia-induced fission and protects cardiomyocytes from cell death decreasing MPTP opening [71•]. Similar results had been described using a chemical inhibitor of Drp1 (mdivi) in an ischemia-reperfusion [72] and in a TAC model [73]. In both systems, mdivi protects from cell death and ameliorates TAC-induced hypertrophy. These results underscore both the relevance of mitochondrial dynamics in cardiomyocyte survival as well as our limited understanding of the regulatory processes beneath. Figure 1c depicts the principal changes in mitochondrial dynamics proteins associated with cardiac remodeling.
A recent development, which certainly will provide interesting answers, is the discovery of microRNA regulation of mitochondrial dynamics. In the normal heart, miR-499 is highly expressed, inhibiting Drp1 activation and mitochondrial fission. Cardiac specific overexpression of miR-499 decreases ischemia-reperfusion myocardial injury and inhibits pathologic remodeling decreasing collagen deposition [74]. Conversely, cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and ultimately heart failure, likely due to its ability to decrease PPAR γ signaling, a known regulator of mitochondrial function. Even more interestingly, the use of an antagomiR-27b attenuated TAC-induced hypertrophy [75]. Several lines of work are currently aimed at clarifying the intricate pathways of microRNA regulation, which may provide a valuable tool for future pharmacological interventions.
Conclusion
Mitochondrial function is critical for normal cardiomyocyte function. Derangements in mitochondrial dynamics, ROS signaling or mitochondrial Ca2+ handling have been associated with cardiac hypertrophy, heart failure and ischemia/reperfusion injury. The identification of suitable therapeutic targets to revert mitochondrial dysfunction associated with heart disease is an ongoing challenge that hopefully will help to alleviate the burden of the cardiovascular pandemic.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–209.
Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure. J Am Coll Cardiol. 2006;48:A56–6.
Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–51.
Parra V, Verdejo H, del Campo A, Pennanen C, Kuzmicic J, Iglewski M, Hill JA, Rothermel BA, Lavandero S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J Bioenerg Biomembr. 2011;43:47–51.
Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198–200.
Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107:15565–70.
Kwon S. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:615–21.
• Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265–13. This paper provides evidence of the regulation of mitophagy and mitochondrial-induced apoptosis via the JNK/FOXO3a pathway in heart failure, revealing a promising therapeutic target in the interference of JNK signal transduction.
Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA, Lavandero S. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244–11.
Vacek TP, Vacek JC, Tyagi SC. Mitochondrial mitophagic mechanisms of myocardial matrix metabolism and remodelling. Arch Physiol Biochem. 2012;118:31–42.
Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B. PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antiox Redox Signal. 2010;13:1011–22.
Huss JM. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–55.
Webster BR, Lu Z, Sack MN, Scott I. The role of sirtuins in modulating redox stressors. Free Rad Biol Med. 2012;52:281–90.
Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–71.
Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011;108:9572–7.
Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35.
Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002;30:2817–24.
Paradies G. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–9.
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–5.
• Dai DF, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. This is an excellent paper that demonstrates how mitochondrial ROS modulation is able to ameliorate overload-induced cardiac remodeling.
Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn 2nd GW, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and G alpha q overexpression-induced heart failure. Circ Res. 2011;108:837–46.
Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49.
• Heineke J, Ritter O. Cardiomyocyte calcineurin signaling in subcellular domains: From the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol. 2012;52:62–73. This is a comprehensive review of the role of calcineurin in cardiac remodeling.
• Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–73. This review summarizes the role of CaMKII in myocardial remodeling.
• Liu Q, Molkentin JD. Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol. 2011;51:474–8. This paper provides an up-to-date review of the role of protein kinase C on heart failure and explores potential pharmacological approaches in heart failure treatment.
Saito S. Beta-adrenergic pathway induces apoptosis through calcineurin activation in cardiac myocytes. J Biol Chem. 2000;275:34528–33.
Salas MA, Valverde CA, Sánchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG, et al. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48:1298–306.
Dong S, Teng Z, Lu FH, Zhao YJ, Li H, Ren H, Chen H, Pan ZW, Lv YJ, Yang BF, et al. Post-conditioning protects cardiomyocytes from apoptosis via PKCε-interacting with calcium-sensing receptors to inhibit endo(sarco)plasmic reticulum–mitochondria crosstalk. Mol Cell Biochem. 2010;341:195–206.
Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–18.
Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–16.
Territo PRP, Mootha VKV, French SAS, Balaban RSR. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol. 2000;278:C423–35.
Moreno-Sánchez R. Contribution of the translocator of adenine nucleotides and the ATP synthase to the control of oxidative phosphorylation and arsenylation in liver mitochondria. J Biol Chem. 1985;260:12554–60.
• Cardenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–83. This paper elegantly demonstrates that constitutive Ca 2+ transfer to the mitochondria is critical for regulating mitochondrial oxidative metabolism and ATP production.
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA. 1999;96:13807–12.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5.
Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010;1797:907–2.
Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, Hajnóczky G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell. 2010;39:121–32.
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell. 2010;38:280–90.
Hayashi T, Rizzuto R, Hajnóczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–8.
García-Pérez C, Hajnóczky G, Csordás G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem. 2008;283:32771–80.
Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–82.
Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998–5007.
Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem. 2006;281:28058–67.
Gupta A, Akki A, Wang Y, Leppo MK, Chacko VP, Foster DB, Caceres V, Shi S, Kirk JA, Su J, et al. Creatine kinase-mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J Clin Invest. 2012;122:291–302.
Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–9.
Di Lisa F, Fan CZ, Gambassi G, Hogue BA, Kudryashova I, Hansford RG. Altered pyruvate dehydrogenase control and mitochondrial free Ca2+ in hearts of cardiomyopathic hamsters. Am J Physiol. 1993;264:H2188–97.
Liu T, O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–88.
Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK, Aranguiz P, Chiong M, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2011;93:320–9.
Zhang Y, Yuan M, Bradley KM, Dong F, Anversa P, Ren J. Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension. 2012;59:680–93.
Chen H. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200.
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–66.
Zheng M, Xiao RP. Role of mitofusin 2 in cardiovascular oxidative injury. J Mol Med. 2010;88:987–91.
Iglewski M, Hill JA, Lavandero S, Rothermel BA. Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hypertens Rep. 2010;12:418–25.
• Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010;88:16–29. This paper provides a thorough review of the regulation of mitochondrial dynamics and its participation on cardiovascular disease.
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–61.
Hu F, Liu F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal. 2011;23:1528–33.
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–28.
Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Härtel S, Jaimovich E, Zorzano A, Hidalgo C, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008;77:387–97.
Neuspiel M. Activated mitofusin 2 signals mitochondrial fusion, interferes with bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–70.
Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143–52.
Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–42.
• Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, et al. The role of nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3:88ra55. This is the first paper to describe a role of a protein involved in ER-mitochondrial coupling in the pathogenesis of pulmonary hypertension.
Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez AE, Pedrozo Z, Chiong M, Parra V, Quest AF, Rothermel BA, et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell Biol. 2012;44:16–20.
• Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–9. This seminal work is the first to describe the involvement of mitochondrial dynamics in heart failure. Recent papers have further expanded these initial results.
Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 2011;90:234–42.
Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh LH, Nicolas V, Alavi MV, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94:408–17.
Kane LA, Youle RJ. Mitochondrial fission and fusion and their roles in the heart. J Mol Med. 2010;88:971–9.
Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, et al. A mutation in the mitochondrial fission gene dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000.
Javadov S, Rajapurohitam V, Kilić A, Hunter JC, Zeidan A, Said Faruq N, Escobales N, Karmazyn M. Expression of mitochondrial fusion–fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol. 2010;106:99–109.
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–8.
• Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–22. This important work was the first to describe a pharmacological strategy to modulate mitochondrial dynamics in an ischemia/reperfusion model.
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in bax/bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204.
Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (mdivi) ameliorates pressure overload induced heart failure. PLoS One. 2012;7:e32388.
Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–8.
Wang J, Song Y, Zhang Y, Xiao H, Sun Q, Hou N, Guo S, Wang Y, Fan K, Zhan D, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Res. 2012;22:516–27.
Acknowledgments
This research was funded in part by Comision Nacional de Ciencia y Tecnologia (CONICYT), Chile: Fondo Desarrollo en Areas Prioritarias 15010006 (S.L.), Anillo de Investigación de Ciencia y Tecnología ACT1111 (S.L., P.F.C., L.G.), FONDECYT 1120212 (S.L.), FONDECYT 1090727 (P.F.C.), FONDECYT 3110114 (R.T.), FONDECYT 3110039 (Z.P.) and FONDECYT 3120220 (C.Q.). We thank the PhD fellowships from MECESUP and CONICYT, Chile to H.E.V., and A.d.C, respectively.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Verdejo, H.E., del Campo, A., Troncoso, R. et al. Mitochondria, Myocardial Remodeling, and Cardiovascular Disease. Curr Hypertens Rep 14, 532–539 (2012). https://doi.org/10.1007/s11906-012-0305-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-012-0305-4