
Abstract
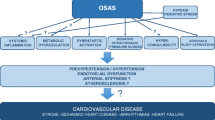
Obstructive sleep apnea (OSA) is increasingly recognized as a novel cardiovascular risk factor. OSA is implicated in the pathogenesis of hypertension, left ventricular dysfunction, coronary artery disease and stroke. OSA exerts its negative cardiovascular consequences through its unique pattern of intermittent hypoxia. Endothelial dysfunction, oxidative stress, and inflammation are all consequences of OSA directly linked to intermittent hypoxia and critical pathways in the pathogenesis of cardiovascular disease in patients with OSA. This review will discuss the known mechanisms of vascular dysfunction in patients with OSA and their implications for cardiovascular disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea (OSA) is a respiratory disorder of sleep characterized by recurrent episodes of complete or partial upper airway obstruction. OSA has an estimated prevalence of 9–24% in middle-aged individuals [1, 2] and is increasingly emerging as a cardiovascular risk factor [3–6]. Several etiological factors in OSA overlap with those of cardiovascular diseases creating difficulty in distinguishing the direct cardiovascular consequences of OSA from its role in exacerbating concomitant cardiovascular disease. Nevertheless, an independent role for OSA in cardiovascular morbidity and mortality is now well supported [4–6].
This review will discuss the pathophysiological responses to episodes of obstructive apnea and hypopnea. These responses include sympathetic activation, increased respiratory workload, and intermittent hypoxia in the immediate term. Endothelial dysfunction, oxidative stress, and inflammation are long-term consequences that mediate cardiovascular disease in patients with OSA. A subsequent review in this series will attempt to present the background and evidence for a causative relationship between OSA and cardiovascular disease with focus on hypertension and heart failure.
Presentation and definition of OSA
The term Sleep Disordered Breathing (SDB) encompasses all types of respiratory disturbance during sleep: obstructive, central, and mixed. Typically, patients have predominance of either central or obstructive events, so SDB is divided broadly into two main clinical syndromes, central and obstructive sleep disorders.
In normal conditions, a tenuous balance between constrictor and dilator forces maintains the patency of the upper airway during sleep [7, 8]. Obstructive events occur when this balance shifts toward the constricting forces [9]. One of the important collapsing factors leading to constriction of the upper airway is the extra-luminal pressure from the tissue surrounding the airway [10], a common condition in obesity.
The presence of compatible clinical symptoms, including excessive daytime sleepiness, and at least five obstructive respiratory events, apneas or hypopneas, per hour of sleep defines Obstructive Sleep Apnea Syndrome. Obstructive apneas result from complete collapse of the upper airway resulting in cessation of airflow against which the inspiratory effort persists. Obstructive hypopneas result from a partial collapse of the upper airway causing reduction in, but not cessation of airflow, and are associated with increased respiratory effort.
The most effective treatment for OSA is continuous positive airway pressure (CPAP), which acts as pneumatic splint keeping the airway open during sleep. Discussion of treatment modalities for OSA is elsewhere in this special issue.
The physiological response to episodes of obstructive apnea and hypopnea
A typical patient with OSA may experience anywhere from five to well over one hundred apnea or hypopnea events per hour. Each of these obstructive respiratory events results in an episode of hypoxia. Re-oxygenation occurs when the episode is terminated by an arousal that restores the airway patency (Fig. 1). The recurrence of these respiratory events and their respective recovery phases produces a characteristic pattern of nocturnal intermittent hypoxia that is unique to OSA. Generally, both apnea and hypopnea events produce the same pattern of intermittent hypoxia. Each episode of hypoxia stimulates the carotid chemoreceptors resulting in sympathetic nerve activation [11] and subsequent surge in blood pressure [12]. As a result, patients with OSA spend their sleep period in a state of intermittent hypoxia and a cycling pattern of recurrent surges of sympathetic activity and blood pressure.

A fragment from a full night sleep study recording (polysomnography) of a patient with severe OSA. Channels from top: LOC: left eye electrooculogram, ROC: right eye electrooculogram, Chin EMG: Electromyogram of the chin, C3-A2 and O2-A1: recordings of two Electroencephalogram leads used for scoring sleep; EKG: Electrocardiogram, Air flow: measured by nasal pressure cannula; Abdomen and chest effort measured by respiratory inductance plesythmography belts, SaO2: Pulse oximetry. Note the recurrent episodes of cessation of flow with persistent respiratory effort (obstructive apneas). Each episode is associated with hypoxia, and is terminated by an arousal and subsequent restoration of the patency of the airway and airflow
Significant experimental evidence has emerged indicating that intermittent hypoxia is a unique physiological state with a profile of biological consequences that is distinct from other types of hypoxia [13–16]. More importantly, intermittent hypoxia is the critical element accounting for most of the immediate and long-term cardiovascular consequences of OSA including hypertension [17–19].
Intermittent hypoxia, sympathetic activation, and the pathogenesis of hypertension
Patients with OSA experience recurrent episodes of sympathetic activation and blood pressure surges throughout the sleep period [20]. This sympathetic activation, along with the increased blood pressure persists during the daytime indicating a link between OSA and the pathogenesis of hypertension [20]. Several studies attempted to explain this blood pressure relation to apnea. Xie et al. reported that a short (20 min) exposure to hypoxia in healthy humans resulted in substantial increase in sympathetic nerve activity, which remained elevated 20 min after withdrawal of the chemical stimulus [21]. In humans exposed to intermittent hypoxia, intact sympathetic pathway was required for the hypertensive response to voluntary apnea [12, 21]. In other experiments, the same investigators, as well as others, confirmed that intermittent hypoxia, and not the respiratory effort associated with apnea, is responsible for the sympathetic activation following episodes of obstructive apnea [11, 22]. Additionally, hypoxia, and not hypercapnea, was critical for the persistence of sympathetic activation following episodes of apnea [23]. Withdrawal of the inhibitory vagal signal associated with inspiration during breath holds was not important for the sympathetic activation and blood pressure surge [24]. In summary, these human experiments confirmed that intermittent hypoxia is the critical stimulus for OSA-associated sympathetic activation [22] and surge in blood pressure following obstructive episodes [12, 21]. The sympathetic response to intermittent hypoxia is associated with a carryover effect in which sympathetic activation and blood pressure surge persist after hypoxia has resolved [25].
Animal models confirmed the role of intermittent hypoxia-induced sympathetic activation in OSA-related hypertension. In a landmark experiment, Brooks et al. developed a dog model of OSA in which they mimicked the upper airway occlusion of OSA. Again, intermittent hypoxia was the mandatory stimulus for the blood pressure response [19]. Fletcher et al. developed an animal model of OSA in which rats were exposed to a protocol of intermittent hypoxia designed to simulate the pattern of nocturnal hypoxia in OSA [26]. An increase in blood pressure occurred in the rats exposed to this intermittent hypoxia protocol compared to control animals [27]. Carotid body denervation prevented the increase in arterial blood pressure. Additionally, either chemical or surgical sympathectomy prevented the blood pressure response to intermittent hypoxia [17, 28]. Similar to human experiments, these series of experiments demonstrated that chemoreception-induced sympathetic activation mediated the blood pressure response to intermittent hypoxia. In particular, intact renal artery, and medullary sympathetic activity were required for the hypertensive response in this animal model [29]. Moreover, in this rat model, intermittent hypoxia not only resulted in increased basal sympathetic activity, but also facilitated enhanced sympathetic response to subsequent episodes of hypoxia [30–32]. Other investigators, using a similar rat model, confirmed that intermittent hypoxia induces long-term facilitation in the sympathetic activation via an effect on the carotid chemoreceptors [25].
Sympathetic overactivity appears to be the critical link between OSA and hypertension [20, 33]. The mechanism by which sympathetic activation contributes to the pathogenesis of hypertension in patients with OSA is not yet fully understood. Parallels do exist in the current understanding of the pathogenesis of essential hypertension, in which sympathetic activity is central [34, 35]. Increased sympathetic tone exerts systemic changes that promote the persistence of elevated blood pressure [36, 37] and augment the response to subsequent sympathetic stimuli [38]. Young patients with early essential hypertension have increased cardiac sympathetic tone compared to age matched controls [39]. In a population-based study, increased heart rate, a manifestation of sympathetic activation, correlated with future development of hypertension [40]. The sympathetic interaction with the renin-angiotensin system may be another important element in the pathogenesis of hypertension [36, 41, 42]. In turn, angiotensin II potentiates the vasoconstrictor effects of sympathetic activation via post-ganglionic effects [43–45]. In the previously mentioned rat model of intermittent hypoxia, Fletcher et al. showed that intermittent hypoxia-induced hypertension was mediated by renal sympathetic nerve activity [17, 46] and that intact renin-angiotensin system was critical for this blood pressure response to intermittent hypoxia [47].
Another important link between OSA and hypertension is the resetting of the baroreflex. Patients and animal models of hypertension demonstrate changes in their autonomic regulation of blood pressure (baroreflex) consistent with adaptation of the baroreceptors to a higher blood pressure set point [48, 49]. This adaptation was reported in patients with OSA both with and without changes in the sensitivity of the baroreflex [50, 51]. Adaptation of the baroreflex in hypertension requires reactive oxygen species (ROS) [52]. Also, long-term facilitation of sympathetic activation in animal models of intermittent hypoxia required ROS [53], establishing another important link with OSA, that is oxidative stress.
Finally, sympathetic activation-mediated vasoconstriction may induce long lasting structural changes in resistance vessels that contribute to the persistence of hypertension [54]. Animal models of intermittent hypoxia demonstrate early structural and functional changes [55], along with impaired vasodilator response to hypoxia [56]. These local effects of increased sympathetic tone on the vascular wall and structure may be mediated by endothelial factors. In the rat model of intermittent hypoxia, endothelin-1 was critical for the sustained increase in blood pressure in response to intermittent hypoxia [57, 58].
In summary, sympathetic activation is central to the pathogenesis of hypertension in OSA. Intermittent hypoxia-induced sympathetic activation and blood pressure increases in patients with OSA persist through the day and mediate a cascade of changes that set the stage for persistent hypertension, probably similar to the conditions of initial stages of essential hypertension [33].
Respiratory effort and the mechanical consequences of OSA
When an obstructive apnea occurs, an increase in the respiratory effort against the closed airway ensues. This inspiratory effort is a result of increased respiratory drive stimulated by the associated hypoxia [59] and results in a profound increase in negative intrathoracic pressure with each inspiration. Interest in the mechanical effects of this negative pressure on cardiac function has been long present. However, the available data suggest that hypoxia and not the respiratory effort is responsible for most of the cardiovascular response to respiratory events [60]. Nevertheless, the effect of this respiratory effort may be more important in patients with existing cardiac dysfunction [61, 62] than in otherwise healthy individuals with OSA. Negative intrathoracic pressure augments the gradient between the intraventricular pressure and the intrathoracic pressure resulting in increased left ventricular work and wall stress during systole [61]. Also, this negative intrathoracic pressure may affect the balance of forces governing the transudation of fluid into the interstitial space resulting in pulmonary edema [63]. Finally, increased venous return to the right ventricle is likely [64], which may cause an increase in preload. Alternatively, some sources suggest that the negative intrathoracic pressure may cause a reduction in venous return and preload and subsequently would reduce stroke volume [61]. It is well established that patients with heart failure and OSA experience immediate improvement in their cardiac work index with elimination of OSA events [65, 66].
Endothelial dysfunction
Endothelial dysfunction generally denotes impairment in endothelium-dependent vasodilation, a function mediated by nitric oxide. Endothelial dysfunction is an important vascular abnormality that precedes the clinical manifestations of cardiovascular disease including hypertension [67, 68]. Dysfunction promotes atherosclerotic changes and arterial lesion development with subsequent clinical complications [69]. Flow-mediated dilation (FMD) is nitric oxide-dependent vasodilation [70] that results from shear-mediated activation of endothelial nitric oxide synthesis in response to an acute increase in blood flow [71]. Measurement of flow-mediated dilation by non-invasive methods provides an assessment of endothelial function and can help in the evaluation of cardiovascular risk [72].
Endothelial dysfunction was demonstrated repeatedly in patients with OSA and in animal models of intermittent hypoxia providing an important link between OSA and cardiovascular diseases. Kato et al. described impaired endothelial-mediated vasodilation in a group of newly diagnosed patients with OSA compared to matched controls [73]. Later, Ip et al. evaluated flow-mediated dilation in a group of OSA patients who were otherwise free of clinically known cardiovascular disease. These investigators also found baseline impairment in endothelial function, which improved after treatment of OSA [74]. A correlation existed between the apnea hypopnea index and the impairment in flow-mediated dilation in both studies. A similar correlation between baseline vascular diameter and oxygen desaturation index was also reported in a large population based study of patients with sleep apnea and cardiovascular disease further supporting a cause-effect relationship [75].
In animal models of intermittent hypoxia, endothelial dysfunction occurred without a change in the levels of endothelial nitric oxide synthase (eNOS) [76]. To date, however, the levels and function of eNOS have not been directly measured in patients with OSA. Circulating levels of nitric oxide (NO) in patients with OSA were reduced at baseline and improved with treatment with CPAP [77]. Oxidative stress plays a major role in disorders of endothelial dysfunction and NO bioavailability [78–81]. Recently, two important studies demonstrated an improvement in endothelial dysfunction in patients with OSA with antioxidant treatment [82, 83], suggesting a similar role for oxidative stress in the mechanism of reduced NO availability in patients with OSA. This provides parallels to other cardiovascular diseases in which oxidative stress-induced endothelial dysfunction is important [84, 85].
Several mechanisms have been proposed to explain the oxidative stress-mediated reduction in NO in patients with OSA. Hypoxia-mediated reduction in molecular oxygen, a substrate of eNOS, in the endothelial cell is one possible mechanism. The increase in free radical production in OSA may cause superoxide-mediated scavenging of NO generating peroxynitrite. Svatikova et al. measured circulating nitrotyrosine as an indicator of peroxynitrite formation in the vascular environment in humans with OSA and found no increase in nitrotyrosine levels [86]. This study, however, does not rule out the accumulation of peroxynitrite in the endothelial cells. Tetrahydrobiopterin (BH4) is a cofactor critical for NO production by eNOS [87, 88]. When this cofactor is depleted in conditions of increased oxidative stress, eNOS produces superoxide instead of NO resulting in endothelial dysfunction [89]. Ascorbate is suggested to replete BH4 [90]. In a relevant study, Grebe et al. showed an improvement in endothelial dysfunction in OSA patients after supplementation with vitamin C, lending support to this pathway [82, 83]. Sources of ROS in the vascular environment are numerous and include mitochondria, xanthine oxidoreductase (XOR), NADPH oxidase, eNOS, Cytochrome P450 enzymes, and the arachidonic acid pathway enzymes lipoxygenase and cyclooxygenase. ROS generated from xanthine oxidoreductase activity during ischemia reperfusion injury [91, 92] are implicated in endothelial dysfunction [93, 94] and hypertension [95, 96]. XOR inhibitors have already been shown to improve endothelial function in humans with other forms of endothelial dysfunction [97–99]. Recent evidence also shows improvement in endothelial dysfunction with xanthine inhibitor treatment in patients with OSA [83].
Asymmetrical dimethylarginine (ADMA) and NG-monomethyl-l-arginine(l-NMMA) are structural analogues of l-Arginine, a substrate for eNOS, and can function as competitive inhibitors for eNOS when their levels accumulate in the vascular environment. Only one human study so far suggests a change in the level of ADMA in patients with OSA with treatment. This reduction of ADMA correlated with the improvement in FMD in these OSA patients [100]. Figure 2 summarizes some of the mechanisms of oxidative stress-mediated endothelial dysfunction that may play a role in endothelial dysfunction in patients with OSA.

A schema of potential mechanisms of oxidative stress-mediated endothelial dysfunction. IH: intermittent hypoxia, ADMA: asymmetrical dimethlarginine, BH4: tetrahydrobiopterin, eNOS: endothelial nitric oxide synthase, NO: nitric oxide, O2: oxygen ONOO-: peroxynitrite
Intermittent hypoxia and oxidative stress
Oxidative stress describes an imbalance between the production of ROS and the antioxidant capacity of a biological system. Oxidative stress occurs in conditions of ischemia reperfusion typical to many disease states. Oxidative stress can be assessed directly by measurement of ROS in biological systems, or indirectly by measurement of oxidation products such as lipids, proteins, or DNA. Additionally, increased oxidative stress can be quantified by measuring the available in vitro antioxidant capacity of a biological system as an indicator of existing oxidative stress. Given the significant influence of environmental factors on oxidative activity, large sample sizes, very meticulous techniques, and sophisticated measurements are usually required to evaluate the role of oxidative stress in a particular disease process [101].
The resemblance between the pattern of intermittent hypoxia associated with OSA and ischemia reperfusion patterns leads to the postulation that OSA would also be associated with oxidative stress. Potential mechanisms for oxidative stress in OSA may be related directly to intermittent hypoxia in a fashion similar to ischemia reperfusion injury, or indirectly via inflammatory response. The increased sympathetic tone and elevated catecholamine levels, a hallmark of OSA, might also be associated with increased ROS production.
Despite the biological plausibility, several earlier studies provided conflicting information regarding the presence of increased oxidative stress in patients with OSA [102–105]. In some of these studies, the negative results may have been due to inadequate controlling, small sample size or use of less-refined techniques than what is available currently [102–104]. Recent studies, particularly ones that involved larger numbers of patients, were able to demonstrate that OSA is indeed associated with increased markers of oxidative stress. Lavie et al. [106] measured plasma levels of thiobarbituric reactive substances (TBARS), a marker of lipid peroxidation, and the levels of paraxonase-1, a marker of antioxidant capacity, in 114 OSA patients and a group of normal controls. The investigators found an increase in lipid peroxidation and a reduction in antioxidant capacity in patients with OSA, which correlated with the severity of their OSA, and subsequently improved with treatment. Similarly, Barcelo et al. [107] found increased lipid peroxidation (oxidized LDL) in patients with OSA compared to controls. Treatment with CPAP reduced the susceptibility of LDL to oxidation.
Several studies evaluated direct or indirect measurements of increased ROS production in patients with OSA. Christou et al., in a series of experiments, evaluated the presence of oxidative stress in blood samples of patients with OSA [108–110]. In one experiment, they measured levels of Diacron reactive oxygen metabolism (D-ROM). Diacron indicates the ability of metals to catalyze the formation of free radicals in the presence of peroxide. They found increased levels of reactive oxygen metabolites which correlated with the severity of OSA [108]. In a later study, the same group found a reduction in this measure of oxidative stress (D-ROM) after treatment with CPAP. Carpagnano et al. found increased levels of 8-isoprostane in exhaled breath condensate and blood of patients with OSA compared to controls [111, 112]. Isoprostane is another measure of lipid peroxidation that may be linked to the pathogenesis of atherosclerosis [113–115]. Tan et al. recently described a link between the lipid abnormality in OSA and atherosclerosis. These investigators found that HDL was dysfunctional in preventing LDL oxidation in patients with OSA [116]. Other investigators evaluated products of oxidized DNA as a measure of increased ROS production. These investigators found increased oxidized DNA products in patients with OSA which correlated with the desaturation index [117]. Takahashi et al. evaluated thioredoxin levels in patients with OSA. Thioredoxin is a protein that is released from cells in response to oxidative stress and may be implicated in myocardiac injury [118] and atherosclerosis. The investigators found increased levels of thioredoxin and a correlation between these levels and severity of OSA [119].
Other studies evaluated cellular antioxidant capacity in patients with OSA. This antioxidant capacity can change in the presence of significant oxidative load and is a potential measurement or marker of existing oxidative stress in the system. Using Trolox Equivalent Antioxidant Capacity assay, Christou et al. found that the antioxidant capacity in the blood of patients with severe OSA was reduced in comparison to normal controls [109]. Another study also found that the antioxidant capacity of the serum in 47 patients with OSA was reduced compared to normal controls and improved with treatment of OSA [120]. Together, these studies indicated an impairment in the protective system from oxidative stress in patients with sleep apnea.
In summary, oxidative stress in patients with OSA is central to the cardiovascular morbidity of OSA. Most recent studies in patients with OSA and animal models of intermittent hypoxia confirm that OSA is associated with oxidative stress, which generally correlated with the severity of sleep apnea, and improved with treatment. The conflicting results of some of the earlier human studies are likely a result of methodology or control of patient variables. Meticulous controlling for environmental and circadian factors along with the controlling of subjects is required for evaluation of oxidative stress in OSA patients. The mechanism of increased oxidative stress in patients with OSA and its consequences remains incompletely understood. Oxidative stress provides an important link in understanding the cardiovascular consequences of OSA. Reactive oxygen species are required for the memory effect of the sympathetic activation in animal models of intermittent hypoxia. Increased oxidative stress in the vascular milieu is involved in the pathogenesis of endothelial dysfunction [82, 121]. Furthermore, cognitive impairment [122, 123], inflammation [124, 125], atherosclerosis [116], hypertension [126]and myocardial injury [118] may all be direct consequences of the oxidative stress in OSA.
OSA and inflammation
A link between OSA and inflammation is an intriguing and increasingly likely component of the pathophysiology of OSA. Several studies suggested that systemic inflammation may be involved in the increased ROS production in OSA [111, 124]. Schulz et al. [124] reported a marked increase in neutrophil superoxide generation in OSA patients when compared to controls. Enhanced superoxide generation by neutrophils decreased with CPAP treatment. The neutrophil chemokines, IL-8 and granulocyte chemotactic protein-2, were significantly higher in OSA patients compared to healthy controls [105].
Htoo et al. assessed nuclear factor kappa B (NF-kappaB) activity in OSA patients compared to control subjects. They determined that neutrophils in OSA patients demonstrate several fold increase in NF-kappaB binding activity compared with control subjects. There was a positive correlation between the degree of NF-kappaB activation and indices of OSA severity. CPAP treatment decreased neutrophil NF-kappaB activation to control levels [127].
In an animal model of OSA, Nácher and colleagues determined that recurrent airway obstruction leads to rapid endothelial cell activation. They noted endothelial cell activation and systemic leukocyte recruitment in the microcirculation, with the apnea group having significantly increased flux of leukocyte activation when compared with the sham groups. P-selectin, an adhesion molecule found in endothelial cells and activated platelets, which plays an essential role in leukocyte recruitment, was up-regulated only in the apnea group [128].
Other studies established that patients with OSA have elevated levels of tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine that plays an important role in neutrophil activation [129, 130]. Vgontzas et al. also demonstrated that Interleukin-6 (IL-6), another pro-inflammatory cytokine, was elevated in OSA patients compared to normal controls. The primary factor influencing TNF-α levels was the degree of sleep disturbance, and the main factor affecting IL-6 levels was body mass index (BMI) [129]. In a recent study, patients with OSA were found to have elevated serum levels of neopterin, a pro-inflammatory marker for macrophage activation, which plays a role in the pathogenesis of cardiovascular disease. In this study, the elevated levels of neopterin also correlated with the severity of the underlying severity of sleep apnea and with the degree of sleep disruption [131].
Expanding literature connecting both IL-6 and C-Reactive Protein (CRP) to OSA is very important, as both of these inflammatory markers are risk factors for cardiovascular disease, including atherosclerosis and coronary heart disease [132–137]. Shamsuzzaman et al. reported that plasma CRP levels were significantly higher in OSA patients compared to age and weight matched controls. In their study, multivariate analysis demonstrated that CRP levels were independently associated with the severity of OSA [138]. In a study assessing adolescents (ages 13–18 years, and free of known cardiovascular disease), an AHI ≥ 5 was associated with increased levels of CRP. The authors concluded that OSA in adolescents confers additional cardiovascular risk beyond that of obesity [139]. Another study examining 69 men who were free of cardiovascular disease demonstrated a strong association between the severity of OSA and CRP levels [131]. Monocyte production of IL-6 was higher in patients with OSA compared to obese control subjects. In those patients with OSA, the factors influencing CRP levels were OSA severity and BMI, and the factors affecting IL-6 levels were BMI and nocturnal hypoxia. Treatment with nasal CPAP significantly decreased levels of CRP and production of IL-6 [140].
Activated leukocytes play an important role in the inflammatory response to injury resulting from hypoxia/reoxygenation that may set off the atherogenic processes [141]. Dyugovaskaya et al. investigated the link between certain adhesion molecules expression on leukocytes and their ability to generate ROS in OSA patients. They found that OSA was associated with increased expression of the adhesion molecules CD15 and CD11c by monocytes, increased adherence of monocytes in culture to human endothelial cells, and increased intracellular ROS production in some monocyte and granulocyte subpopulations. Nasal CPAP reversed most of these inflammatory activities. Minoguchi et al. examined carotid intima-media thickness (IMT) along with inflammatory markers associated with cardiovascular disease (CRP, IL-6, and IL-18). Carotid IMT correlated with serum CRP levels, IL-6, and IL-18, duration of OSA-related hypoxia, and severity of OSA. The primary factor influencing carotid IMT was duration of hypoxia during total sleep time [142]. These findings indicate that patients with OSA are exposed to atherogenic insult nightly [143].
Therefore, OSA appears increasingly linked to cardiovascular morbidity via a distinct inflammatory response. This response is complex and includes several humoral and cellular pathways that are only minimally understood so far. This inflammatory response directly links OSA with the pathogenesis of atherosclerosis.
Summary
In otherwise healthy individuals, OSA constitutes a significant risk factor for the development of cardiovascular disease or the progression of existent cardiovascular disorders toward heart failure, stroke, or death. OSA exerts its negative cardiovascular consequences through its unique pattern of intermittent hypoxia. Endothelial dysfunction, oxidative stress, and inflammation are all consequences of OSA directly linked to intermittent hypoxia and critical pathways in the pathogenesis of cardiovascular disease in patients with OSA.
References
Young T, Skatrud J, Peppard PE (2004) Risk factors for obstructive sleep apnea in adults. J Am Med Assoc 291(16):2013–2016. doi:10.1001/jama.291.16.2013
Young T et al (1993) The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328(17):1230–1235. doi:10.1056/NEJM199304293281704
McNicholas WT, Bonsigore MR (2007) Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 29(1):156–178. doi:10.1183/09031936.00027406
Campos-Rodriguez F et al (2005) Mortality in obstructive sleep apnea-hypopnea patients treated with positive airway pressure. Chest 128(2):624–633. doi:10.1378/chest.128.2.624
Doherty LS et al (2005) Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest 127(6):2076–2084. doi:10.1378/chest.127.6.2076
Marin JM et al (2005) Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365(9464):1046–1053
Badr MS (1996) Effect of ventilatory drive on upper airway patency in humans during NREM sleep. Respir Physiol 103(1):1–10. doi:10.1016/0034-5687(95)00079-8
Jordan AS, White DP (2008) Pharyngeal motor control and the pathogenesis of obstructive sleep apnea. Respir Physiol Neurobiol 160(1):1–7. doi:10.1016/j.resp.2007.07.009
Malhotra A et al (2001) Upper-airway collapsibility: measurements and sleep effects. Chest 120(1):156–161. doi:10.1378/chest.120.1.156
Horner RL et al (1989) Sites and sizes of fat deposits around the pharynx in obese patients with obstructive sleep apnoea and weight matched controls. Eur Respir J 2(7):613–622
Morgan BJ, Denahan T, Ebert TJ (1993) Neurocirculatory consequences of negative intrathoracic pressure vs. asphyxia during voluntary apnea. J Appl Physiol 74(6):2969–2975
Katragadda S et al (1997) Neural mechanism of the pressor response to obstructive and nonobstructive apnea. J Appl Physiol 83(6):2048–2054
Peng YJ, Prabhakar NR (2003) Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol 94(6):2342–2349
Prabhakar NR et al (2001) Intermittent hypoxia: cell to system. Am J Physiol Lung Cell Mol Physiol 281(3):L524–L528
Prabhakar NR, Kline DD (2002) Ventilatory changes during intermittent hypoxia: importance of pattern and duration. High Al Med Biol 3(2):195–204. doi:10.1089/15270290260131920
Cutler MJ et al (2004) Periods of intermittent hypoxic apnea can alter chemoreflex control of sympathetic nerve activity in humans. Am J Physiol Heart Circ Physiol 287(5):H2054–H2060. doi:10.1152/ajpheart.00377.2004
Lesske J et al (1997) Hypertension caused by chronic intermittent hypoxia—influence of chemoreceptors and sympathetic nervous system. J Hypertens 15(12 Pt 2):1593–1603
Fletcher EC (2001) Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol 90(4):1600–1605
Brooks D et al (1997) Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 99(1):106–109. doi:10.1172/JCI119120
Somers VK et al (1995) Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96(4):1897–1904. doi:10.1172/JCI118235
Xie A et al (2000) Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol 89(4):1333–1339
Cutler MJ et al (2004) Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol 96(2):754–761. doi:10.1152/japplphysiol.00506.2003
Xie A et al (2001) Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol 91(4):1555–1562
Khayat RN et al (2004) Role of sensory input from the lungs in control of muscle sympathetic nerve activity during and after apnea in humans. J Appl Physiol 97(2):635–640. doi:10.1152/japplphysiol.00241.2004
Prabhakar NR et al (2005) Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32(5–6):447–449. doi:10.1111/j.1440-1681.2005.04209.x
Fletcher EC et al (1992) Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol 72(5):1978–1984
Fletcher EC et al (1992) Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension 19(6 Pt 1):555–561
Fletcher EC et al (1992) Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 20(5):612–619
Bao G et al (1997) Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol 83(1):95–101
Dick TE et al (2007) Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92(1):87–97. doi:10.1113/expphysiol.2006.035758
Sica AL et al (2000) Chronic-intermittent hypoxia: a model of sympathetic activation in the rat. Respir Physiol 121(2–3):173–184. doi:10.1016/S0034-5687(00)00126-2
Greenberg HE et al (1999) Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol 86(1):298–305
Fletcher EC (2003) Sympathetic over activity in the etiology of hypertension of obstructive sleep apnea. Sleep 26(1):15–19
Julius S, Esler MD, Randall OS (1975) Role of the autonomic nervous system in mild human hypertension. Clin Sci Mol Med Suppl 2:243s–252s
Esler M et al (1986) Mechanism of elevated plasma noradrenaline in the course of essential hypertension. J Cardiovasc Pharmacol 8(Suppl 5):S39–S43
Esler M et al (1976) High-renin essential hypertension: adrenergic cardiovascular correlates. Clin Sci Mol Med Suppl 3:181s–184s
Oparil S, Zaman MA, Calhoun DA (2003) Pathogenesis of hypertension. Ann Intern Med 139(9):761–776
Guo GB, Abboud FM (1984) Angiotensin II attenuates baroreflex control of heart rate and sympathetic activity. Am J Physiol 246(1 Pt 2):H80–H89
Rumantir MS et al (2000) The ‘adrenaline hypothesis’ of hypertension revisited: evidence for adrenaline release from the heart of patients with essential hypertension. J Hypertens 18(6):717–723. doi:10.1097/00004872-200018060-00009
Kim JR et al (1999) Heart rate and subsequent blood pressure in young adults: the CARDIA study. Hypertension 33(2):640–646
Farsang C et al (1981) Effect of prazosin and oxprenolol on plasma renin activity and blood pressure in patients with essential hypertension. Cardiology 67(3):164–171
Winternitz SR, Katholi RE, Oparil S (1980) Role of the renal sympathetic nerves in the development and maintenance of hypertension in the spontaneously hypertensive rat. J Clin Invest 66(5):971–978. doi:10.1172/JCI109966
Ma X et al (2006) Dual mechanisms of angiotensin-induced activation of mouse sympathetic neurones. J Physiol 573(Pt 1):45–63. doi:10.1113/jphysiol.2006.106716
Ma X, Abboud FM, Chapleau MW (2001) A novel effect of angiotensin on renal sympathetic nerve activity in mice. J Hypertens 19(3 Pt 2):609–618. doi:10.1097/00004872-200103001-00014
Ma X et al (2001) Angiotensin selectively activates a subpopulation of postganglionic sympathetic neurons in mice. Circ Res 88(8):787–793. doi:10.1161/hh0801.089542
Fletcher EC, Bao G, Li R (1999) Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension 34(2):309–314
Fletcher EC, Orolinova N, Bader M (2002) Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J Appl Physiol 92(2):627–633
Somers VK, Mark AL, Abboud FM (1991) Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest 87(6):1953–1957. doi:10.1172/JCI115221
Chapleau MW, Hajduczok G, Abboud FM (1988) Mechanisms of resetting of arterial baroreceptors: an overview. Am J Med Sci 295(4):327–334. doi:10.1097/00000441-198804000-00019
Cooper VL et al (2007) Daytime variability of baroreflex function in patients with obstructive sleep apnoea: implications for hypertension. Exp Physiol 92(2):391–398. doi:10.1113/expphysiol.2006.035584
Brooks D et al (1999) Baroreflex control of heart rate in a canine model of obstructive sleep apnea. Am J Respir Crit Care Med 159(4 Pt 1):1293–1297
Li Z et al (1996) Oxygen-derived free radicals contribute to baroreceptor dysfunction in atherosclerotic rabbits. Circ Res 79(4):802–811
Peng YJ et al (2003) Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100(17):10073–10078. doi:10.1073/pnas.1734109100
Rouwet EV et al (2002) Hypoxia induces aortic hypertrophic growth, left ventricular dysfunction, and sympathetic hyperinnervation of peripheral arteries in the chick embryo. Circulation 105(23):2791–2796. doi:10.1161/01.CIR.0000017497.47084.06
Phillips SA et al (2006) Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J Appl Physiol 100(4):1117–1123. doi:10.1152/japplphysiol.00994.2005
Phillips SA et al (2004) Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol 286(1):H388–H393. doi:10.1152/ajpheart.00683.2003
Allahdadi KJ, Walker BR, Kanagy NL (2005) Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension 45(4):705–709. doi:10.1161/01.HYP.0000153794.52852.04
Kanagy NL, Walker BR, Nelin LD (2001) Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension 37(2 Part 2):511–515
Sforza E et al (1996) Role of chemosensitivity in intrathoracic pressure changes during obstructive sleep apnea. Am J Respir Crit Care Med 154(6 Pt 1):1741–1747
Chen L, Scharf SM (1997) Comparative hemodynamic effects of periodic obstructive and simulated central apneas in sedated pigs. J Appl Physiol 83(2):485–494
Chen L, Shi Q, Scharf SM (2000) Hemodynamic effects of periodic obstructive apneas in sedated pigs with congestive heart failure. J Appl Physiol 88(3):1051–1060
Hall MJ et al (1998) Magnitude and time course of hemodynamic responses to Mueller maneuvers in patients with congestive heart failure. J Appl Physiol 85(4):1476–1484
Fletcher EC et al (1999) Pulmonary edema develops after recurrent obstructive apneas. Am J Respir Crit Care Med 160((5 Pt 1)):1688–1696
Bradley TD, Floras JS (2003) Sleep apnea and heart failure: Part I: obstructive sleep apnea. Circulation 107(12):1671–1678. doi:10.1161/01.CIR.0000061757.12581.15
Naughton MT (1998) Impact of treatment of sleep apnoea on left ventricular function in congestive heart failure. Thorax 53(Suppl 3):S37–S40
Tkacova R et al (1998) Effects of continuous positive airway pressure on obstructive sleep apnea and left ventricular afterload in patients with heart failure. Circulation 98(21):2269–2275
Koller A, Huang A (1994) Impaired nitric oxide-mediated flow-induced dilation in arterioles of spontaneously hypertensive rats. Circ Res 74(3):416–421
Brevetti G et al (2003) Endothelial dysfunction and cardiovascular risk prediction in peripheral arterial disease: additive value of flow-mediated dilation to ankle-brachial pressure index. Circulation 108(17):2093–2098. doi:10.1161/01.CIR.0000095273.92468.D9
Ross R (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362(6423):801–809. doi:10.1038/362801a0
Juonala M et al (2007) Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in young Finns study. Circulation 116(12):1367–1373. doi:10.1161/CIRCULATIONAHA.107.690016
Ungvari Z et al (2002) Impaired nitric oxide-mediated flow-induced coronary dilation in hyperhomocysteinemia: morphological and functional evidence for increased peroxynitrite formation. Am J Pathol 161(1):145–153
Corretti MC et al (2002) Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol 39(2):257–265. doi:10.1016/S0735-1097(01)01746-6
Kato M et al (2000) Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 102(21):2607–2610
Ip MS et al (2004) Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 169(3):348–353. doi:10.1164/rccm.200306-767OC
Nieto FJ et al (2004) Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. Am J Respir Crit Care Med 169(3):354–360. doi:10.1164/rccm.200306-756OC
Tahawi Z et al (2001) Altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol 90(5):2007–2013, discussion 2000
Ip MS et al (2000) Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am J Respir Crit Care Med 162(6):2166–2171
Geny B et al (2006) Comments on point-counterpoint “flow-mediated dilation does/does not reflect nitric oxide-mediated endothelial function”. J Appl Physiol 100(1):362. doi:10.1152/japplphysiol.01313.2005
Wolin MS et al (1998) Oxidant—nitric oxide signalling mechanisms in vascular tissue. Biochemistry. Biokhimiia 63(7):810–816
Ogita H, Liao J (2004) Endothelial function and oxidative stress. Endothelium 11(2):123–132. doi:10.1080/10623320490482664
Touyz RM, Schiffrin EL (2004) Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol 122(4):339–352. doi:10.1007/s00418-004-0696-7
Grebe M et al (2006) Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am J Respir Crit Care Med 173(8):897–901. doi:10.1164/rccm.200508-1223OC
El Solh AA et al (2006) Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Respir J 27(5):997–1002
Pieper GM, Dembny K, Siebeneich W (1998) Long-term treatment in vivo with NOX-101, a scavenger of nitric oxide, prevents diabetes-induced endothelial dysfunction. Diabetologia 41(10):1220–1226. doi:10.1007/s001250051055
Katayama Y et al (2004) Oral vitamin C ameliorates smoking-induced arterial wall stiffness in healthy volunteers. J Atheroscler Thromb 11(6):354–357
Svatikova A et al (2004) Circulating free nitrotyrosine in obstructive sleep apnea. Am J Physiol Regul Integr Comp Physiol 287(2):R284–R287. doi:10.1152/ajpregu.00241.2004
Cosentino F et al (2008) Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart (British Cardiac Society) 94(4):487–492. doi:10.1136/hrt.2007.122184
Channon KM (2004) Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med 14(8):323–327. doi:10.1016/j.tcm.2004.10.003
Stroes E et al (1997) Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 99(1):41–46. doi:10.1172/JCI119131
Heller R et al (1999) L-Ascorbic acid potentiates nitric oxide synthesis in endothelial cells. J Biol Chem 274(12):8254–8260. doi:10.1074/jbc.274.12.8254
Zweier JL et al (1994) Measurement and characterization of free radical generation in reoxygenated human endothelial cells. Am J Physiol 266(3 Pt 1):C700–C708
Zweier JL (1998) Free radical generation in human endothelial cells exposed to anoxia and reoxygenation. Transplant Proc 30(8):4228–4232. doi:10.1016/S0041-1345(98)01399-2
Houston M et al (1999) Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J Biol Chem 274(8):4985–4994. doi:10.1074/jbc.274.8.4985
Ohara Y, Peterson TE, Harrison DG (1993) Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest 91(6):2546–2551. doi:10.1172/JCI116491
Mervaala EM et al (2001) Endothelial dysfunction and xanthine oxidoreductase activity in rats with human renin and angiotensinogen genes. Hypertension 37(2 Part 2):414–418
Berry CE, Hare JM (2004) Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 555(Pt 3):589–606. doi:10.1113/jphysiol.2003.055913
Butler R et al (2000) Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 35(3):746–751
Farquharson CA et al (2002) Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 106(2):221–226. doi:10.1161/01.CIR.0000022140.61460.1D
Guthikonda S et al (2004) Role of xanthine oxidase in conduit artery endothelial dysfunction in cigarette smokers. Am J Cardiol 93(5):664–668. doi:10.1016/j.amjcard.2003.11.046
Ohike Y et al (2005) Amelioration of vascular endothelial dysfunction in obstructive sleep apnea syndrome by nasal continuous positive airway pressure—possible involvement of nitric oxide and asymmetric NG, NG-dimethylarginine. Circ J 69(2):221–226. doi:10.1253/circj.69.221
Suzuki YJ et al (2006) Oxidative stress and oxidant signaling in obstructive sleep apnea and associated cardiovascular diseases. Free Radic Biol Med 40(10):1683–1692. doi:10.1016/j.freeradbiomed.2006.01.008
Svatikova A et al (2005) Oxidative stress in obstructive sleep apnoea. Eur Heart J 26(22):2435–2439. doi:10.1093/eurheartj/ehi440
Wali SO et al (1998) Susceptibility of LDL to oxidative stress in obstructive sleep apnea. Sleep 21(3):290–296
Ozturk L et al (2003) Lipid peroxidation and osmotic fragility of red blood cells in sleep-apnea patients. Clin Chim Acta 332(1–2):83–88. doi:10.1016/S0009-8981(03)00126-8
Alzoghaibi MA, Bahammam AS (2005) Lipid peroxides, superoxide dismutase and circulating IL-8 and GCP-2 in patients with severe obstructive sleep apnea: a pilot study. Sleep Breath 9(3):119–126. doi:10.1007/s11325-005-0022-1
Lavie L, Vishnevsky A, Lavie P (2004) Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 27(1):123–128
Barcelo A et al (2000) Abnormal lipid peroxidation in patients with sleep apnoea. Eur Respir J 16(4):644–647. doi:10.1034/j.1399-3003.2000.16d13.x
Christou K et al (2003) Reactive oxygen metabolites (ROMs) as an index of oxidative stress in obstructive sleep apnea patients. Sleep Breath 7(3):105–110. doi:10.1007/s11325-003-0105-9
Christou K et al (2003) Antioxidant capacity in obstructive sleep apnea patients. Sleep Med 4(3):225–228. doi:10.1016/S1389-9457(02)00253-8
Christou K et al (2008) Nasal continuous positive airway pressure treatment reduces systemic oxidative stress in patients with severe obstructive sleep apnea syndrome. Sleep Med (in press). doi:10.1016/j.sleep.2007.10.011
Carpagnano GE et al (2002) Increased 8-isoprostane and interleukin-6 in breath condensate of obstructive sleep apnea patients. Chest 122(4):1162–1167. doi:10.1378/chest.122.4.1162
Carpagnano GE et al (2003) 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest 124(4):1386–1392. doi:10.1378/chest.124.4.1386
Tangirala RK et al (2001) Reduction of isoprostanes and regression of advanced atherosclerosis by apolipoprotein E. J Biol Chem 276(1):261–266. doi:10.1074/jbc.M003324200
Pratico D (1999) F(2)-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis 147(1):1–10. doi:10.1016/S0021-9150(99)00257-9
Pratico D et al (1997) Localization of distinct F2-isoprostanes in human atherosclerotic lesions. J Clin Invest 100(8):2028–2034. doi:10.1172/JCI119735
Tan KC et al (2006) HDL dysfunction in obstructive sleep apnea. Atherosclerosis 184(2):377–382. doi:10.1016/j.atherosclerosis.2005.04.024
Yamauchi M et al (2005) Oxidative stress in obstructive sleep apnea. Chest 127(5):1674–1679. doi:10.1378/chest.127.5.1674
Park AM, Suzuki YJ (2007) Effects of intermittent hypoxia on oxidative stress-induced myocardial damage in mice. J Appl Physiol 102(5):1806–1814. doi:10.1152/japplphysiol.01291.2006
Takahashi K et al (2008) Plasma thioredoxin, a novel oxidative stress marker, in patients with obstructive sleep apnea before and after nasal continuous positive airway pressure. Antioxid Redox Signal 10(4):715–726. doi:10.1089/ars.2007.1949
Barcelo A et al (2006) Antioxidant status in patients with sleep apnoea and impact of continuous positive airway pressure treatment. Eur Respir J 27(4):756–760. doi:10.1183/09031936.06.00067605
Teramoto S et al (2007) Improvement of endothelial function with allopurinol may occur in selected patients with OSA: effect of age and sex. Eur Respir J 29(1):216–217, author reply 217–218. doi:10.1183/09031936.00104806
Veasey SC et al (2004) Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep 27(2):194–201
Xu W et al (2004) Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience 126(2):313–323. doi:10.1016/j.neuroscience.2004.03.055
Schulz R et al (2000) Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162(2 Pt 1):566–570
Row BW et al (2004) Platelet-activating factor receptor-deficient mice are protected from experimental sleep apnea-induced learning deficits. J Neurochem 89(1):189–196. doi:10.1111/j.1471-4159.2004.02352.x
Troncoso Brindeiro CM et al (2007) Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol 293(5):H2971–H2976. doi:10.1152/ajpheart.00219.2007
Htoo AK et al (2006) Activation of nuclear factor kappaB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep Breath 10(1):43–50. doi:10.1007/s11325-005-0046-6
Nacher M et al (2007) Recurrent obstructive apneas trigger early systemic inflammation in a rat model of sleep apnea. Respir Physiol Neurobiol 155(1):93–96. doi:10.1016/j.resp.2006.06.004
Vgontzas AN et al (1997) Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J Clin Endocrinol Metab 82(5):1313–1316. doi:10.1210/jc.82.5.1313
Kataoka T et al (2004) The effect of surgical treatment of obstructive sleep apnea syndrome on the plasma TNF-alpha levels. Tohoku J Exp Med 204(4):267–272. doi:10.1620/tjem.204.267
Punjabi NM et al (2007) Elevated levels of neopterin in sleep-disordered breathing. Chest 132(4):1124–1130. doi:10.1378/chest.07-0743
Yudkin JS et al (2000) Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 148(2):209–214. doi:10.1016/S0021-9150(99)00463-3
Ridker PM et al (2000) Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 101(15):1767–1772
Lindmark E et al (2001) Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy. J Am Med Assoc 286(17):2107–2113. doi:10.1001/jama.286.17.2107
Haverkate F et al (1997) Production of C-reactive protein and risk of coronary events in stable and unstable angina. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. Lancet 349(9050):462–466. doi:10.1016/S0140-6736(96)07591-5
Lindahl B et al (2000) Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during instability in coronary artery disease. New Engl J Med 343(16):1139–1147. doi:10.1056/NEJM200010193431602
Burke AP et al (2002) Elevated C-reactive protein values and atherosclerosis in sudden coronary death: association with different pathologies. Circulation 105(17):2019–2023. doi:10.1161/01.CIR.0000015507.29953.38
Shamsuzzaman AS et al (2002) Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 105(21):2462–2464. doi:10.1161/01.CIR.0000018948.95175.03
Larkin EK et al (2005) Variation of C-reactive protein levels in adolescents: association with sleep-disordered breathing and sleep duration. Circulation 111(15):1978–1984. doi:10.1161/01.CIR.0000161819.76138.5E
Yokoe T et al (2003) Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 107(8):1129–1134. doi:10.1161/01.CIR.0000052627.99976.18
Lefer AM (1999) Role of the beta2-integrins and immunoglobulin superfamily members in myocardial ischemia-reperfusion. Ann Thorac Surg 68(5):1920–1923. doi:10.1016/S0003-4975(99)01017-6
Minoguchi K et al (2005) Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med 172(5):625–630. doi:10.1164/rccm.200412-1652OC
Dyugovskaya L, Lavie P, Lavie L (2002) Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 165(7):934–939
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khayat, R., Patt, B. & Hayes, D. Obstructive sleep apnea: the new cardiovascular disease. Part I: obstructive sleep apnea and the pathogenesis of vascular disease. Heart Fail Rev 14, 143–153 (2009). https://doi.org/10.1007/s10741-008-9112-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-008-9112-z