
Abstract
Obstructive sleep apnea (OSA) is a common disorder that has been associated with an increased risk of atherosclerosis and its clinical manifestations. While treatment with continuous positive airway pressure (CPAP) has been shown to exert several beneficial effects on cardiovascular disease and prognosis in observational studies, CPAP was not effective in three recent randomized controlled trials unless it was used for more than 4 hours of sleep.
Zusammenfassung
Die obstruktive Schlafapnoe (OSA) stellt eine häufige Erkrankung dar, die mit einem erhöhten Risiko für Arteriosklerose und deren klinischen Manifestationen in Zusammenhang gebracht wird. In Beobachtungsstudien wurden zwar für die Behandlung mit kontinuierlichem positivem Atemwegsdruck („continuous positive airway pressure“, CPAP) verschiedene günstige Auswirkungen auf Herz-Kreislauf-Erkrankungen und deren Prognose nachgewiesen, in drei aktuellen randomisierten kontrollierten Studien stellte sich die CPAP-Therapie jedoch nicht als wirksam heraus, wenn sie nicht mindestens vier Stunden lang während des Schlafs eingesetzt wurde.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
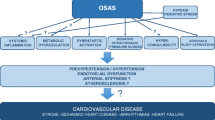
Atherosclerosis describes the narrowing of an artery due to formation of plaques. Depending on where these atherosclerotic plaques appear, they result in different manifestations, such as coronary heart disease, peripheral arterial disease, cerebral arterial disease, and their secondary disorders such as myocardial infarction or stroke. Cardiovascular diseases (CVD) are the leading cause of death in developed countries [1]; 31% of all deaths worldwide are due to CVD [2]. There are several risk factors contributing to the genesis of atherosclerosis. The major risk factors are smoking, hypertension, dyslipidemia, and diabetes. Obstructive sleep apnea (OSA) is highly associated with CVD [3, 4] and cardiovascular events like coronary revascularization, myocardial infarction, stroke, or cardiovascular death.
Obstructive sleep apnea is a sleep disorder where breathing becomes shallow or stops completely, caused by obstruction of the upper airway. It is the most common type of sleep apnea. Estimates of its prevalence vary between 2 and 50%, with a higher prevalence among men and of mild OSA [5, 6]. The prevalence is approximately three to four times higher in populations with CDV than in those without [7]. There is an especially high prevalence among people with resistant hypertension (70–83%), heart failure (>50%), and stroke [7,8,9]. Therefore, screening for OSA is particularly recommended in these individuals.
An apnea is defined as a cessation of breathing for more than 10 s; a hypopnea is a reduction of airflow with an oxygen desaturation of the blood by more than 4% [10]. The number of apnea and hypopnea episodes per hour define the apnea–hypopnea index (AHI). OSA is defined as AHI ≥ 5/h plus daytime sleepiness, or AHI ≥ 15/h. The degree of its severity is defined as follows: AHI 5–14.9/h defines mild, 15–29.9/h moderate, and ≥30/h severe OSA [10, 11]. Due to the cessation of airflow, oxygen desaturations occur until the apnea is ended by a so-called arousal, a short interruption of sleep [12, 13]. The frequent alternations of hypoxemia and arousals lead to sleep fragmentation. These are accompanied by activation of the sympathetic nerve system, oxidative stress, systemic inflammation, and intrathoracic pressure swings, which lead to progression of atherosclerotic lesions. Arousals do not compulsorily lead to complete awakening but make sleep less restorative and cause the blood pressure to rise [13]. The combination of OSA with its typical accompanying symptom, daytime sleepiness, is called obstructive sleep apnea syndrome (OSAS). Daytime sleepiness is often assessed by questionnaires, such as the Epworth Sleepiness Scale (ESS) [3]. A test result of ≥11 points indicates significant daytime sleepiness.
Further symptoms include snoring, headache, and memory deficits, up to symptoms of dementia. As one can imagine, by causing severe daytime sleepiness, falling asleep several times a day, and possibly even the imposition of a driving ban, OSAS can have a serious effect on people’s lives. All in all, patients with OSAS suffer from poor sleep and life quality [5].
Pathomechanisms linking OSA and atherosclerosis
Obstructive sleep apnea, particularly when severe, is strongly associated with CVD such as coronary heart disease, atrial fibrillation, congestive heart failure, stroke, and peripheral arterial disease [3, 7, 12,13,14,15]. All these diseases share common risk factors, such as arterial hypertension or metabolic syndrome.
As OSA is often attended by the abovementioned comorbidities, which are also risk factors for atherosclerosis, it is difficult to evaluate the exclusive impact of OSA on atherosclerosis. Nevertheless, there are several pathophysiological changes in OSA supposed to have a direct influence on atherosclerosis [6], as demonstrated in numerous animal and human studies. The apneic events in OSA lead to intermittent hypoxemia, sleep fragmentation, and intrathoracic pressure swings, which are considered as main links between OSA and atherosclerosis [4, 14].
Intermittent hypoxemia and sleep deprivation seem to contribute to dyslipidemia, systemic inflammation, neuronal nerve activity, and oxidative stress [7, 15,16,17,18]. Furthermore, the reactive oxygen species formed induce systemic inflammation and endothelial dysfunction, further promoting atherosclerosis [17, 19]. The idea of hypoxemia contributing to formation of atherosclerotic lesions is additionally supported by findings showing that the nightly amount of hypoxemia is associated with CVD [20]. Beyond that, it induces new and contributes to existing atherosclerotic lesions in mouse models [15].
Furthermore, there are hemodynamic and mechanistic changes in the thorax during an apnea. The unsuccessful attempts to breath against the closed upper airway lead to activation of the sympathetic nerve system, negative intrathoracic pressure, and thus an increased left ventricular transmural gradient and an increased left ventricular afterload, together causing diastolic dysfunction, a rise in arterial blood pressure, and shear stress of the arterial wall [4, 21]. The hemodynamic changes lead to a rise in blood pressure and renal hypoperfusion, as shown in a pig model study. Here, an obstructive apnea was imitated in pigs and a negative intrathoracic pressure was thus built up. Heart rate, blood pressure, renal blood flow, and the neurohumoral response (plasma renin activity, plasma aldosterone concentration, urinary protein/creatinine ratio) were quantified. The measurements were repeated after renal denervation. The repeated tracheal occlusions caused a rise in blood pressure, reduced renal perfusion, and induced neurohumoral changes. These changes could be attenuated by renal denervation, while hypoxia and hypercapnia did not change. Therefore, the authors concluded that the effects on blood pressure and the neurohumoral response were mainly mediated by the sympathetic drive, rather than by the blood gases alone [22].
Additionally, snoring itself has been identified as possibly influencing endothelial functions in a ventilated rabbit model [23]. The vibration is assumed to be transmitted through the thorax up to the arterial wall, thus decreasing vasodilatation [24].
The activation of the sympathetic nerve system in OSA has been verified in further animal and human models. For example, a direct measurement of neuronal activity via neurogram in rats and microneurography in humans has been performed. The sympathetic activation leads to vasoconstriction and a higher blood pressure and heart rate even during daytime [18, 25].
In addition to the vascular, inflammatory, hemodynamic, mechanical, and neural pathological pathways, there is also a metabolic one. There is a known correlation between OSA and metabolic syndrome [28]. Metabolic syndrome is a combination of obesity, hypertension, impaired glucose tolerance, and dyslipidemia, resulting in a predisposition for atherosclerosis. Several pathophysiological reactions caused by OSA—e. g., the increase in sympathetic hormones and elevation of inflammatory markers like tumor necrosis factor (TNF)-alpha, C‑reactive protein (CRP), or interleukin (IL)-6—lead to both increased insulin resistance and atherosclerosis. These pathologies not only seem to coexist, but may also have synergistic effects [28] that possibly further increase the risk of atherosclerosis.
The current German S3 guidelines for “Sleeping Disorders—Sleep-Related Abnormal Breathing” state that an association of OSA with atherosclerosis is probable but not proven [3]. The abovementioned processes that connect OSA with atherosclerosis are explanations for a direct influence on the one hand, and common pathologic pathways on the other. Both are closely related and hard to separate. All in all, it is ultimately unclear whether OSA itself causes atherosclerosis, although the results of present research clearly imply that it does.
The effect of CPAP therapy on atherosclerosis
Continuous positive airway pressure (CPAP) is the standard therapy for OSA and a class I recommendation [11] for moderate to severe OSA (AHI ≥ 15/h) and mild OSA (AHI ≥ 5/h) with daytime sleepiness or cardiovascular comorbidities [3, 4]. Several cross-sectional and longitudinal studies have shown that OSA is associated with elevated blood pressure, one of the main risk factors for atherosclerosis [29, 30]. Furthermore, OSA is associated with CVD as a consequence of atherosclerosis [12,13,14,15,16, 21, 26, 31]. OSA is seen as a modifiable atherosclerotic risk factor, and its treatment may help to reduce cardiovascular complications [14]. CPAP therapy creates, as its name implies, a continuous positive airway pressure, and thereby functions as a splint for the upper airway. It prevents airway collapse and, accordingly, the intrathoracic pressure swings and cycles of hypoxemia and arousals. This affects the pathophysiology in several beneficial ways.
For many of the abovementioned mediators promoting the atherosclerotic process in OSA, a positive effect of CPAP therapy has been investigated in smaller studies and has been reviewed repeatedly [4, 5, 14].
Patients with heart failure profit from the improved hemodynamic situation under CPAP therapy. This effect is explained, for instance, by an increased cardiac output due to the reduction of afterload and an improvement of diastolic function. Both right and left ventricular function can improve [30]. The effects on, e. g., blood pressure, were shown to occur immediately, which promotes the idea of the direct effect on the hemodynamic situation under the given therapy [31].
Furthermore, positive airway pressure (PAP) therapy has been shown to attenuate sympathetic nerve activity [25, 30]. Some studies reveal that CPAP even reduces atherosclerosis itself, as measured by carotid artery intima–media thickness (IMT), which is a well-accepted predictor for cardiovascular risk and a surrogate marker for early stages of atherosclerosis [32]. These effects could not be reproduced in randomized controlled trials [33]. However, a recent meta-analysis showed that the effects on IMT are significant in subgroups with more severe OSA and longer-term CPAP use [33].
As to its effect on the reduction of arterial hypertension, findings are inconsistent. Some studies do not support the notion of CPAP therapy lowering the blood pressure significantly, particularly in nonsleepy patients [27, 34, 35]. A meta-analysis of randomized controlled trials showed a significant reduction in arterial hypertension in sleepy OSA patients, with increasing effects with longer nightly CPAP use and a higher baseline AHI [28]. Two meta-analyses of five respectively six randomized controlled trials showed a significant reduction of blood pressure in resistant hypertension, with a larger effect than that reported in OSA patients without resistant hypertension [36, 37]. OSA is typically linked to resistant, nocturnal, and nondipper hypertension [38]. Nondippers seem to have an even higher incidence of cardiovascular events, and CPAP therapy is presumably more effective in treating resistant hypertension [39].
Severe OSA (AHI ≥ 30/h) is independently associated with cardiovascular events and cardiovascular death in both men and women [29, 40]. Observational studies support the hypothesis that CPAP therapy reduces cardiovascular events [29, 40,41,42,43], but due to the lack of large randomized controlled trials, it is not finally clear whether patients really profit from CPAP therapy in terms of cardiovascular morbidity and mortality. To date, three larger randomized controlled trials have been performed to approach this question. These are summarized in Table 1.
The multicenter randomized SAVE study included more than 1300 patients with a diagnosis of coronary artery disease or cerebrovascular disease. Therapy with CPAP did not prevent cardiovascular events in patients with moderate to severe OSA and established CVD [36]. It should be noted that in this patient population, the average duration of CPAP therapy was only 3.3 h per night, but there was also no significant difference in the subgroup analysis of adherent patients compared to the control group [36]. There was indeed, however, a significant reduction in daytime sleepiness (quantified by ESS), accidents with injury, and improvement in quality of life.
The RICCADSA trial, also published in 2016, examined whether asymptomatic patients with mild to moderate OSA benefit from CPAP treatment. In this monocentric randomized controlled trial, 244 CAD patients (shortly after coronary revascularization) with OSA (AHI > 15/h) without daytime sleepiness were included. Patients were randomized into two groups with either autoCPAP or no ventilation therapy. The combined primary endpoint comprised a further coronary revascularization, myocardial infarction, stroke, and cardiovascular death. The primary endpoint did not differ significantly between the two groups [44]. Thus, the treatment of asymptomatic OSA with CPAP therapy did not seem to improve the cardiovascular outcome of the intention-to-treat group. However, patients seemed to profit from application for more than 4 h per night and a good compliance [44]. It is debatable whether this is solely achieved by the CPAP treatment or generally better compliance and health consciousness. Similar results were shown by Barbé et al.: CPAP therapy did not lead to a significant reduction in arterial hypertension or cardiovascular events [27].
A recent meta-analysis comparing ten randomized controlled trials, including the SAVE study and RICCADSA trial, failed to show a reduction in cardiovascular events or cardiovascular death. As possible reasons, low adherence among the patients and a relatively small number of clinical endpoints occurring were discussed [14]. The effect of CPAP therapy on nonsleepy OSA patients with acute coronary syndrome (ACS) will be evaluated in the ISAAC trial, which has not yet been finally published [46]. The actual data concentrate on secondary cardiovascular prevention. The patients enrolled in the abovementioned studies already suffered from CAD; the knowledge of CPAP therapy as a primary prevention is limited. This may be interesting to investigate, as atherosclerosis is a chronic disease developing over the course of time and patients may profit from an earlier intervention.
Conclusion
OSA is associated with atherosclerosis and consequently with CVD. It is not entirely clear to which extent OSA causes atherosclerosis itself, and how much is simply due to common pathologic pathways. Nevertheless, there are many vascular, inflammatory, neuronal, and mechanical changes occurring in OSA which promote the formation of atherosclerotic plaques. CPAP has been shown to attenuate several of these pathological changes.
Considering the insights offered by recent research, CPAP treatment is recommended for patients with symptomatic OSA, as it improves quality of life, daytime sleepiness, cognition, and wellbeing. CPAP treatment for OSA in nonsleepy patients for cardiovascular risk reduction cannot be recommended for all patients at present. However, patients with better compliance seem to profit from CPAP therapy, and one may speculate that more hours of CPAP application result in improved sleep quality, quality of life, blood pressure, and thus even in fewer cardiovascular events. Therefore, further randomized controlled trials with bigger sample sizes are required to definitively identify subgroups or special phenotypes which may profit from CPAP therapy and to evaluate its long-term effects on atherosclerosis. We should be aware that cardiovascular comorbidities are frequent in OSA patients, and an optimal treatment of cardiovascular risk factors should be aimed for in all OSA patients.
References
WHO (2018) Global health estimates 2016: deaths by cause, age, sex, by country and by region, 2000–2016. World Health Organization, Geneva
WHO (2017) WHO Fact sheet N°317. www.who.int/mediacentre/factsheets/fs317/en/ (Created 05.2017). Accessed 29 Oct 2018
Riemann D, Baum E, Cohrs S et al (2017) S3-Leitlinie Nicht erholsamer Schlaf/Schlafstörungen. Somnologie 21(2). https://doi.org/10.1007/s11818-016-0097-x
Skowasch D, Bönner G, Nickenig G (2009) Obstruktive Schlafapnoe als Risikofktor der Arteriosklerose
Maeder MT, Schoch OD, Rickli H (2016) A clinical approach to obstructive sleep apnea as a risk factor for cardiovascular disease. Vasc Health Risk Manag 12:85–103. https://doi.org/10.2147/VHRM.S74703
Tuleta I, Pabst S, Juergens UR, Nickenig G, Skowasch D (2011) Obstructive sleep apnoea as a risk factor for atherosclerosis—implication for preventive and personalised treatment. Epma J 2:39–47. https://doi.org/10.1007/s13167-011-0070-5
Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A et al (2008) Sleep apnea and cardiovascular disease: an American Heart Association/american College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. Circulation 118:1080–1111. https://doi.org/10.1161/CIRCULATIONAHA.107.189375 (In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health))
Logan AG, Perlikowski SM, Mente A, Tisler A, Tkacova R, Niroumand M et al (2001) High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 19:2271–2277
Pearse SG, Cowie MR (2016) Sleep-disordered breathing in heart failure. Eur J Heart Fail 18:353–361. https://doi.org/10.1002/ejhf.492
Mbata G, Chukwuka J (2012) Obstructive sleep apnea hypopnea syndrome. Ann Med Health Sci Res 2:74–77. https://doi.org/10.4103/2141-9248.96943
Oldenburg O, Arzt M, Bitter T et al (2015) Positionspapier „Schlafmedizin in der Kardiologie“. Kardiologe 9(140). https://doi.org/10.1007/s12181-015-0654-8
Eckert DJ, Younes MK (2014) Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol (1985) 116:302–313. https://doi.org/10.1152/japplphysiol.00649.2013
Quan SF (2012) Obstructive sleep apnea and cardiovascular disease: back and forward in time over the last 25 years. Southwest J Pulm Crit Care 5:206–217
Drager LF, McEvoy RD, Barbe F, Lorenzi-Filho G, Redline S, INCOSACT Initiative (International Collaboration of Sleep Apnea Cardiovascular Trialists) (2017) Sleep apnea and cardiovascular disease: lessons from recent trials and need for team science. Circulation 136:1840–1850. https://doi.org/10.1161/CIRCULATIONAHA.117.029400
Song D, Fang G, Greenberg H, Liu SF (2015) Chronic intermittent hypoxia exposure-induced atherosclerosis: a brief review. Immunol Res 63:121–130. https://doi.org/10.1007/s12026-015-8703-8
Adedayo AM, Olafiranye O, Smith D, Hill A, Zizi F, Brown C et al (2014) Obstructive sleep apnea and dyslipidemia: evidence and underlying mechanism. Sleep Breath Schlaf Atm 18:13–18. https://doi.org/10.1007/s11325-012-0760-9
Eisele H‑J, Markart P, Schulz R (2015) Obstructive sleep apnea, oxidative stress, and cardiovascular disease: evidence from human studies. Oxid Med Cell Longev 2015:608438. https://doi.org/10.1155/2015/608438
Xing T, Pilowsky PM (2010) Acute intermittent hypoxia in rat in vivo elicits a robust increase in tonic sympathetic nerve activity that is independent of respiratory drive. J Physiol 588:3075–3088. https://doi.org/10.1113/jphysiol.2010.190454
Lavie L, Lavie P (2009) Molecular mechanisms of cardiovascular disease in OSAHS: the oxidative stress link. Eur Respir J 33:1467–1484. https://doi.org/10.1183/09031936.00086608
Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Nieto FJ et al (2001) Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 163:19–25. https://doi.org/10.1164/ajrccm.163.1.2001008
Lalande S, Luoma CE, Miller AD, Johnson BD (2012) Effect of changes in intrathoracic pressure on cardiac function at rest and during moderate exercise in health and heart failure. Exp Physiol 97:248–256. https://doi.org/10.1113/expphysiol.2011.061945
Linz D, Mahfoud F, Linz B, Hohl M, Schirmer SH, Wirth KJ et al (2014) Effect of obstructive respiratory events on blood pressure and renal perfusion in a pig model for sleep apnea. Am J Hypertens 27:1293–1300. https://doi.org/10.1093/ajh/hpu036
Amatoury J, Howitt L, Wheatley JR, Avolio AP, Amis TC (2006) Snoring-related energy transmission to the carotid artery in rabbits. J Appl Physiol Bethesda Md 1985 100:1547–1553. https://doi.org/10.1152/japplphysiol.01439.2005
Lui MM-S, Sau-Man M (2012) OSA and atherosclerosis. J Thorac Dis 4:164–172. https://doi.org/10.3978/j.issn.2072-1439.2012.01.06
Somers VK, Dyken ME, Clary MP, Abboud FM (1995) Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96:1897–1904. https://doi.org/10.1172/JCI118235
Castaneda A, Jauregui-Maldonado E, Ratnani I, Varon J, Surani S (2018) Correlation between metabolic syndrome and sleep apnea. World J Diabetes 9:66–71. https://doi.org/10.4239/wjd.v9.i4.66
Barbé F, Durán-Cantolla J, Sánchez-de-la-Torre M, Martínez-Alonso M, Carmona C, Barceló A et al (2012) Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: a randomized controlled trial. JAMA 307:2161–2168. https://doi.org/10.1001/jama.2012.4366
Haentjens P, Van Meerhaeghe A, Moscariello A, De Weerdt S, Poppe K, Dupont A et al (2007) The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med 167:757–764. https://doi.org/10.1001/archinte.167.8.757
Marin JM, Carrizo SJ, Vicente E, Agusti AGN (2005) Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet Lond Engl 365:1046–1053. https://doi.org/10.1016/S0140-6736(05)71141-7
Spießhöfer J, Fox H, Lehmann R, Efken C, Heinrich J, Bitter T et al (2016) Heterogenous haemodynamic effects of adaptive servoventilation therapy in sleeping patients with heart failure and Cheyne-Stokes respiration compared to healthy volunteers. Heart Vessels 31:1117–1130. https://doi.org/10.1007/s00380-015-0717-6
Oldenburg O, Bartsch S, Bitter T, Schmalgemeier H, Fischbach T, Westerheide N et al (2012) Hypotensive effects of positive airway pressure ventilation in heart failure patients with sleep-disordered breathing. Sleep Breath Schlaf Atm 16:753–757. https://doi.org/10.1007/s11325-011-0571-4
Amin M (2016) Can continuous positive airway pressure reverse carotid artery atherosclerosis in obstructive sleep apnea? Egypt J Chest Dis Tuberc 65:765–769
Chen L‑D, Lin L, Lin X‑J, Ou Y‑W, Wu Z, Ye Y‑M et al (2017) Effect of continuous positive airway pressure on carotid intima-media thickness in patients with obstructive sleep apnea: a meta-analysis. PLoS ONE 12:e184293. https://doi.org/10.1371/journal.pone.0184293
Becker HF, Jerrentrup A, Ploch T, Grote L, Penzel T, Sullivan CE et al (2003) Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation 107:68–73
Robinson GV, Langford BA, Smith DM, Stradling JR (2008) Predictors of blood pressure fall with continuous positive airway pressure (CPAP) treatment of obstructive sleep apnoea (OSA). Thorax 63:855–859. https://doi.org/10.1136/thx.2007.088096
McEvoy RD, Antic NA, Heeley E, Luo Y, Ou Q, Zhang X et al (2016) CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med 375:919–931. https://doi.org/10.1056/NEJMoa1606599
Iftikhar IH, Valentine CW, Bittencourt LRA, Cohen DL, Fedson AC, Gíslason T et al (2014) Effects of continuous positive airway pressure on blood pressure in patients with resistant hypertension and obstructive sleep apnea: a meta-analysis. J Hypertens 32:2341–2350. https://doi.org/10.1097/HJH.0000000000000372 (discussion 2350)
Torres G, Sánchez-de-la-Torre M, Barbé F (2015) Relationship between OSA and hypertension. Chest 148:824–832. https://doi.org/10.1378/chest.15-0136
Feldstein CA (2016) Blood pressure effects of CPAP in nonresistant and resistant hypertension associated with OSA: a systematic review of randomized clinical trials. Clin Exp Hypertens N Y N 1993 38:337–346. https://doi.org/10.3109/10641963.2016.1148156
Campos-Rodriguez F, Martinez-Garcia MA, de la Cruz-Moron I, Almeida-Gonzalez C, Catalan-Serra P, Montserrat JM (2012) Cardiovascular mortality in women with obstructive sleep apnea with or without continuous positive airway pressure treatment: a cohort study. Ann Intern Med 156:115–122. https://doi.org/10.7326/0003-4819-156-2-201201170-00006
Cassar A, Morgenthaler TI, Lennon RJ, Rihal CS, Lerman A (2007) Treatment of obstructive sleep apnea is associated with decreased cardiac death after percutaneous coronary intervention. J Am Coll Cardiol 50:1310–1314. https://doi.org/10.1016/j.jacc.2007.06.028
Doherty LS, Kiely JL, Swan V, McNicholas WT (2005) Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest 127:2076–2084. https://doi.org/10.1378/chest.127.6.2076
Milleron O, Pillière R, Foucher A, de Roquefeuil F, Aegerter P, Jondeau G et al (2004) Benefits of obstructive sleep apnoea treatment in coronary artery disease: a long-term follow-up study. Eur Heart J 25:728–734. https://doi.org/10.1016/j.ehj.2004.02.008
Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E (2016) Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with Nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 194:613–620. https://doi.org/10.1164/rccm.201601-0088OC
Yu J, Zhou Z, McEvoy RD, Anderson CS, Rodgers A, Perkovic V et al (2017) Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 318:156–166. https://doi.org/10.1001/jama.2017.7967
Esquinas C, Sánchez-de-la Torre M, Aldomá A, Florés M, Martínez M, Barceló A et al (2013) Rationale and methodology of the impact of continuous positive airway pressure on patients with ACS and nonsleepy OSA: the ISAACC Trial. Clin Cardiol 36:495–501. https://doi.org/10.1002/clc.22166
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
L. Biener, C. Pizarro, G. Nickenig, and D. Skowasch declare that they have no competing interests.
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Biener, L., Pizarro, C., Nickenig, G. et al. Obstructive sleep apnea and atherosclerosis—update 2019. Somnologie 23, 3–7 (2019). https://doi.org/10.1007/s11818-019-0194-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11818-019-0194-8
Keywords
- Continuous positive airway pressure
- Cardiovascular diseases
- Hypertension
- Coronary artery disease
- Risk factors