
Abstract
Vicatia thibetica de Boiss.: a herb in the family Apiaceae, has been used for over a hundred years as an essential medicinal and edible plant in the Bai ethnic group of Dali City. However, due to the lack of study on plastid genomes of V. thibetica, studies of comparison and phylogeny with other related species remain scarce. In the current study, we assembled, annotated, and characterized the entire chloroplast (cp) genome of V. thibetica through high-throughput sequencing for the first time, compared with published whole chloroplast genomes from the same family. A phylogenetic analysis of the chloroplast genome has also been performed. The whole chloroplast genome of V. thibetica was 145,796 in size and consisted of a large single-copy region (LSC; 92,186 bp), a small single-copy region (SSC; 17,452 bp), and a pair of inverted repeat regions (IRs; 18,079 bp) forming a circular quadripartite structure. Annotation resulted in 128 genes, including 84 protein-coding genes (PCGs), 35 transfer RNA genes (tRNAs), eight ribosomal genes (rRNAs), and one pseudogene. Repeat sequence analysis displayed V. thibetica plastid genome contains 75 simple repeats, 37 long repeats, and 29 tandem repeats. Compared with the cp genome of other Apiaceae species, a common feature was that the IR regions of the genome were more conservative compared to the LSC and SSC regions. Highly variable hotspots included rps16, ndhC-trnV-UAC, clpP, ycf1, and ndhB in the genomes, which supply valuable molecular markers for phylogeny, identification, and classification in the Apiaceae family. The results of phylogenetic analysis strongly supported the genus Vicatia as an independent genus in the family Apiaceae, in which the closest affinities to the related species of Angelica, Peucedanum, and Ligusticum were observed. In conclusion, the first chloroplast genome of Vicatia reported in this study may improve our understanding of phylogenetic relationship of different genera of Apiaceae. In addition, the current data will be valuable as chloroplast genomic resource for species identification and population genetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vicatia thibetica is a medicinal and edible plant species of the Apiaceae family, which belongs to the Apioideae subfamily (Pu et al. 2005). V. thibetica is distributed mainly in Tibet, Yunnan, and Sichuan provinces of China, growing on hillsides, grasslands, forests, river beaches, at an altitude of 2700–4000 m (She et al. 1979), and have been artificially cultivated in Dali city (Zhou et al. 2007). The dry root of V. thibetica was called Xigui (Zhou et al. 2007), a Bai ethnic medicine with a dual purpose of medicinal and edible use (Zhou et al. 2007; Jiang et al. 2016). Studies have shown that Xigui contained umbelliferone, bergapten, ferulic acid, apigenin, and other chemical components (Zhang et al. 2004). It has different pharmacological effects such as anti-aging, anti-fatigue, anti-dysmenorrhea, anti-oxidation, promoting intelligence, and improving immunity (Dong et al. 2018).
Xigui has been used by Bai folk in Dali City for more than a hundred years and has the efficacy of supplementing blood, invigorating qi, and regulating menstruation (Zhou et al. 2007; Jiang et al. 2016). Interestingly, Xigui was often used to substitute for Chinese herbal medicine Angelica sinensis in Northwest Yunnan and West Sichuan (Zhou et al. 2007). However, previous studies through differences in chemical composition revealed that Xigui could not be substituted for A. sinensis (Zhang et al. 2004). Since the morphological similarity of V. thibetica and A. sinensis makes it challenging to make an accurate distinction in the appearance, a molecular method is urgently needed to distinguish them.
Chloroplast is the site of photosynthesis in green plants and a vital organelle involved in the synthesis of pigments, lipids, hormones, and ribosomes (Raven and Allen 2003). Studying chloroplast genomes is essential for exploring plant molecular markers, the structure of chloroplast DNA, and species relationships (Tang et al. 2011). In addition, the plant cp genomes were characterized by a conserved structure and a high substitution rate and were considered a valuable source for plant molecular identification, genetic diversity assessment, and phylogenetic analysis (Dong et al. 2012, 2014). Park et al. (2019) found that the ycf4-cemA fragment could distinguish the herbal medicine Ligusticum officinale and Angelica polymorpha based on divergent region analysis of chloroplast genome. Zhang et al. (2019a, b) indicated that the chloroplast genome was used as a super-barcode to accurately discriminate various Dracaena species, successfully solving the problem of identifying Dracaena plants.
So far, the NCBI database has included above 5488 chloroplast genome sequences. However, there have been no reports on chloroplast genomes of related species of Vicatia. Therefore, the entire V. thibetica cp genome sequence was obtained by Illumina NovaSeq sequencing in this study. Meanwhile, comparative analysis with the cp genome of other Apiaceae species, including A. sinensis, facilitates this genus’s phylogenetic and molecular identification studies. Moreover, this study provides a basis for the classification, identification, conservation genetics, and resource exploitation of Vicatia plants.
Materials and methods
Plant materials, DNA extraction, and illumina sequencing
Clean, fresh leaves of V. thibetica were obtained from Machang Town, Heqing County, Dali, Yunnan Province, China (100° 20′ 4″ E, 26° 3′ 57″ N; elevation 3010 m) and identified by Professor Cong-long Xia (College of Pharmacy, Dali University). Total DNA was extracted using the E.Z.N.A® Plant DNA kit (OMEGA). Extracted DNA was checked for quality and integrity by 1% agarose gel electrophoresis, followed by concentration and content using TBS380 Picogreen (Invitrogen). Then, DNA extracts were fragmented to 300–500 bp using Covaris M220 sonication. These fragments were then purified using TruSeq™ Nano DNA Sample Prep Kit for the trimming, 3’-end adenylation, and ligation index adaptor. The sequencing library was created by PCR amplification of appropriate size fragments, whose library was sequenced with paired-end (2 × 150 bp) using Illumina NovaSeq 6000 platform (Shanghai Biozeron Biotech Co, Ltd).
Genome assembly and annotation
Raw data for V. thibetica were created with 150 bp paired-end read lengths. Since there will be some data with lower quality in the raw sequencing data, to make the subsequent assembly more accurate, it will be quality sheared using the software Trimmomatic (Bolger et al. 2014).
After that, the use of NOVOPlasty software assembled high-quality reads into contigs (Dierckxsens et al. 2016). Next, clean reads were aligned back onto the scaffold obtained from the assembly, and the assembly results were locally assembled and optimized according to the paired-end and overlap relationship of the reads. Then, the inner holes of the assembly result were repaired using GapCloser v1.12 software.
Finally, the start position of assembled chloroplast sequence was corrected using the reference sequence, and the positions and orientations of the four chloroplast partitions (LSC/IRA/SSC/IRB) were determined, resulting in the ultimate genome sequence. Download the species closest to this species from NCBI as a reference sequence, and the assembly results were annotated using DOGMA (Dual Organellar Genome Annotator) (Wyman et al. 2004). A physical map of the cp genome was mapped in the OGDRAW (Organellar Genome Draw) program (Lohse et al. 2007). Finally, the sequence was submitted to NCBI with accession number MZ189732.
Codon preference analysis
Statistical and preference analyses of amino acid usage frequency and the relative synonymous codon usage (RSCU) of the 84 CDS sequences in the chloroplast genome of V. thibetica were performed using the codonw1.4.2 program (Sharp et al. 1986).
Repeat analysis in V. thibetica chloroplast genome
The SSRs were detected using MISA (Thiel et al. 2003) with the following thresholds: Ten repeat units for mononucleotide SSRs, five units for dinucleotide SSRs, four repeat units for trinucleotide repeat SSRs, and three repeat units for tetranucleotide, pentanucleotide, and hexanucleotide repeat SSRs. The online Tandem Repeats Finder (TRF) v4.04 was used to find tandem repeats (Benson 1999). Using the software REPuter (http://bibiserv.techfak.uni-bielefeld.de/reputer/) for long repeat analysis with parameters set to minimal repeat size of 30 bp, Hamming distance of 3 (Kurtz et al. 2001). Four repeat types, forward repeat (F), palindromic repeat (P), reverse repeat (R), and complement repeat (C), were detected with sequence similarity ≥ 90%.
Comparative genomic analysis of the V. thibetica chloroplast genome
Sequence similarity alignment analysis was performed using the Shuffle-LAGAN model (Brudno et al. 2003) of the mVISTA online program (http://genome.lbl.gov/vista/mvista/submit.shtml) (Frazer et al. 2004). Among them, five published chloroplast genomes (C. paradoxum [MK780227.1], B. chinense [NC_046774.1], L. sinense [NC_038088.1], A. sinensis [MH430891.1], and P. praeruptorumin [MN016968.1]) were selected for pairwise comparison using the plastome of V. thibetica as the reference genome. In addition, the boundaries of chloroplast genomes from nine species of the family Apiaceae were mapped using the IRscope online program (https://irscope.shinyapps.io/irapp/) (Amiryousefi et al. 2018).
Analysis of chloroplast genome by sliding window
V. thibetica was the species of this study, and the remaining eight complete chloroplast genome sequences were downloaded from NCBI. Chloroplast genomes were aligned using MAFFT v.7.129 (Katoh and Standley 2013) and BioEdit software (Hall. 1999). DanSP v 5.1 software was employed to perform sliding window analysis and computed nucleotide diversity index Pi. The step length and window length were set to 200 bp and 600 bp, respectively (Rozas et al. 2017).
Phylogenetic analysis
Phylogenetic inference was performed based on plastid genomes, and the complete chloroplast genome sequences of 37 Apiaceae species were used for phylogenetic tree construction (Table S5). The cp genomes of species E. trifoliatus and A. cordata of the Araliaceae family were set as outgroup. The selected sequences were first aligned using MAFFT v.7.129 (Katoh and Standley 2013) and then manually adjusted with BioEdit (Hall 1999). After that, the aligned sequences were analyzed for phylogenetic reconstruction based on the maximum likelihood method using the IQTree software with 1000 bootstrap replicates (Nguyen et al. 2015). In the Partition Finder V2.1.1 software, the most suitable model of nucleotide substitution was selected using the Bayesian information criterion (BIC) (Lanfear et al. 2012). Reconstruction of the ML tree with the best-fit model TVM + F + R7. Bootstrap values were computed using UFBoo built into IQTree, computed by fast bootstrapping to avoid low support values (Minh et al. 2013). In addition, we also constructed a Neighbor-Joining (NJ) phylogenetic tree by MEGA X software with kimura-2 as the parameter and 1000 bootstrap values.
Results
Composition and characteristics of the chloroplast genome
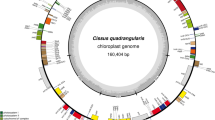
The complete circular four-segment structure of the V. thibetica chloroplast genome was obtained, with a total length of 145,796 bp, consisting of a large single-copy region (LSC, 92,186 bp), a small single-copy region (SSC, 17,452 bp), and a pair of inverted repeat regions (18,079 bp) (Fig. 1). Annotation of the whole genome sequence of V. thibetica resulted in 128 genes, including 37 tRNA genes, eight rRNA genes, 84 protein-coding genes, and one pseudogene ycf1. We observed 14 genes with duplications in the IRs region, containing four PCGs (one gene containing introns), four rRNAs, and six tRNAs (two genes containing introns) (Tables 1, 2). Meanwhile, these genes contained 19 identified introns, of which 11 were PCGs and eight were tRNAs. The ycf3 and clpP were genes that contained two introns, whereas all others contained only a single intron (Table S1). Moreover, total GC content was 37.7% in the V. thibetica plastome, whereas GC content in IR region (44.8%) exceeded that in LSC (36.1%) and SSC (31.1%) regions (Table 1). In addition, the genome was composed of approximately 56.4% of the coding region (82,220 bp) and 43.6% of the non-coding region (63,576 bp). The frequencies of adenine (A), thymine (T), cytosine (C), and guanine (G) in the cp genome of V. thibetica were 44,785, 46,091, 28,055, and 26,864 bp, accounting for 30.7%, 31.6%, 19.2%, and 18.4% of the genome, respectively (Table 1).

Physical map of V. thibetica cp genome. Genes shown inside the circle are transcribed clockwise, and genes outside the circle are transcribed counter-clockwise. Thick black lines represent inverted repeats
In V. thibetica, a total of 84 protein-coding genes encoding 23,508 codons were observed. These included 61 unique codons encoding 20 amino acids and three stop codons (Fig. 2). Of these, 2506 (10.66%) codons encoded leucine and 252 (1.36%) codons encoded cysteine, which were the most and least encoded amino acids, respectively, in the V. thibetica chloroplast genome. Thirty codons with an RSCU value greater than 1 indicated a high preference for codon usage, whereas 32 codons with an RSCU value less than 1 indicated a low preference for usages (Table S2).

Analysis of relative synonymous codon usage (RSCU) in the chloroplast genome of V. thibetica
Comparison with chloroplast genomes of other apiaceae
Comparative analysis based on the nine genera of chloroplast genomes of family Apiaceae (Vicatia, Angelica, Peucedanum, Ligusticum, Semenovia, Heracleum, Bupleurum, Chamaesium, and Hydrocotyle) has never been reported earlier. The overall GC content of these species ranged from 36.7% (S. gyirongensis) to 38.6% (L. sinensis). The size of the V. thibetica cp genome was slightly similar to that of the L. sinense and P. praeruptorum (Wu et al. 2020; Li et al. 2019). In addition, the plastid genome was largest for B. chinense (155,545 bp) and smallest for A. sinensis (142,822 bp). In all species, the length of the SSC region was conservative, ranging from 17,505 bp (B. Chinese) to 18,690 bp (H. sibthorpioides). IR regions varied markedly in length, from 25,108 (A. sinensis) to 52,610 bp (B. chinense). The IR were the smallest in A. sinensis (25,108 bp). The length of the LSC region is highest for A. sinensis (99,964 bp) and shortest for C. paradoxum (84,162 bp).
Except for 134 genes observed in species belonging to the Hydrocotyloideae subfamily (H. sibthorpioide), the total gene numbers in all species were conservative (Table 3). In most species, 129 genes were identified, and eight rRNAs (four rRNA duplications) were detected in each species. V. thibetica, C. paradoxum, B. chinense, and P. praeruptorum contained 84 protein-coding genes. S. gyirongensis, H. sibthorpioides, and H. yungningense contained 85 protein-coding genes, whereas L. sinense contained 87 and A. sinensis contained 83. The V. thibetica, A. sinensis, and P. praeruptorum chloroplast genomes possessed 35 tRNA genes, C. paradoxum, B. chinense, and H. sibthorpioides possessed 37 tRNA genes, whereas the L. sinense, S. gyirongensis, and H. yungningense chloroplast genome possessed 36 tRNA genes (Table 3).
Repeats analysis
Simple sequence repeats (SSRs) are extensively used in species evolution and population genetics studies (Powell et al. 1995; Roullier et al. 2011). We determined the type, distribution, and frequency of simple sequence repeats in the V. thibetica plastome. A total of 75 perfect SSRs were identified by MISA analysis, which included 40 mono-, 22 di-, one tri-, eight tetra-, two penta- and two hexanucleotide repeats (Fig. 3A). In the V. thibetica whole chloroplast genome, 10, 53, and 12 SSRs were present in the IR, LSC, and SSC regions (Fig. 3B). Thus, simple sequence repeats were primarily distributed in the LSC region, which accounted for 71% of the total, whereas IR regions (13%) were least distributed (Fig. 4A). The repeat type that occurred three times was the most frequent in the LSC region, including tetranucleotide, pentanucleotide, hexanucleotide, whereas other frequencies of classified repeat types appeared either once or did not appear in LSC, SSC, and IR regions (Fig. 4B–D). The results showed that mononucleotide motifs accounted for approximately 53% of SSRs. Out of these, mononucleotide A and T repeat units were the most dominant parts (Table S3), accounting for 46.7%.

Analysis of SSR repeat types and numbers in V. thibetica chloroplast genome. A SSRs in the complete cp genome. B SSRs in the LSC, SSC, and IR regions

The type, distribution, and frequency of SSRs in the V. thibetica chloroplast genome. A Distribution of SSRs in the LSC, SSC, and IR regions. B–D Frequencies of repeat motifs in the LSC, SSC, and IR regions
Repetitive sequence utilized illegitimate recombination and slipped strand mismatches play a significant role in genomic rearrangements and mutation (Bausher et al. 2006; Jansen et al. 2007). Twenty-nine tandem repeats were determined in the plastome of V. thibetica (Table S4), which was higher compared to Chamaesium paradoxum (15), Bupleurum chinense (26), Ligusticum sinense (24), and smaller compared to Angelica sinensis (32), and Peucedanum praeruptorum (38). Detected repeats were distributed in intergenic spacer (IGS), protein-coding regions (CDS), and intronic regions, while most tandem repeats were in IGS and intronic regions. Of the tandem repeats detected in the V. thibetica chloroplast genome, 23 were situated in the IGS regions, and two were distributed in the intronic region of trnI-GAU. However, just three and one repeats were presented in CDS regions of ycf2 and ycf1, respectively. The size of these repeats varied from 25 to 98 bp, which was the longest repeat present in the IGS region of rps16/trnQ-UUG.
Long repeats play an essential role in the variation, expansion, and rearrangement of the complete chloroplast genome (Asaf et al. 2018). The following four repeat types were observed by the software REPuter: forward (F), reverse (R), complement (C), palindromic (P). The minimal repeat size is 30 bp for all repeat types. There were 19 F repeats, 16 P repeats, one R repeats, and one C repeats in the cp genome of V. thibetica (Table 4). In all, there were 37 long repeats in V. thibetica plastome. Most of the repeat sizes were between 30 and 39 bp (81.08%), followed by 40–49 bp (16.21%), whereas 50–59 bp were the least (0.03%). Meanwhile, the R and C repeat of the V. thibetica cp genome only contained 30–39 bp (Fig. 5). At the first long repeat position, 83.8% of repeat sequences were observed in non-coding regions. Two repeats were situated in the tRNAs (0.06%), the other five repeats (13.51%) were situated in the PCGs, particularly psaA, psaB, and ycf2 (Table 4).

Long repeat sequences in cp genome of V. thibetica. REPuter was used to identify repeat sequences with a length ≥ 30 bp, and sequences identified ≥ 90% in the chloroplast genomes. F, P, R, and C indicate the repeat types F (forward), P (palindrome), R (reverse), and C (complement). Repeats with different lengths are indicated in different colors
Comparative genomics analysis
This study performed a global alignment of published whole chloroplast genome sequences from five Apiaceae species using the online genome alignment tool mVISTA, while V. thibetica was set as reference sequence (Fig. 6). This result confirmed that the variation in the non-coding region of the six cp genomic sequences was higher compared to that in the conserved protein-coding region. In addition, the LSC region, and the SSC region had significantly higher number of variation compared to the IR regions, whereas the rRNA gene region was highly conservative, with few variants detected. The genes which showed more variations were matK, rpoC2, rpoC1, rpoB, ycf1, ycf2, and ndhF, while others were very highly conserved. Variants in IGS were exceeded in gene regions such as rps16-trnQ-UUG, atpH-atpI, rpoB-trnC-GCA, trnE-UUC-psbD, ndhC-trnV-UAC, rpl16-rps3, ycf4-cemA, and petA-psbJ. As illustrated in Fig. 6, the plastid genome of L. sinensis, A. sinensis, and P. praeruptorum had high similarity with V. thibetica, indicating that they are closely related species. However, the genomes of C. paradoxum and B. chinense presented more variations compared with the reference genome.

Comparing the chloroplast genome sequences of V. thibetica, C. paradoxum, B. chinense, L.sinense, A. sinensis, P. praeruptorum generated with mVISTA. Grey arrows indicate the position and direction of each gene. Red and blue areas show the intergenic and genic regions, respectively. The vertical scale indicates the percentage of identity, ranging from 50 to 100%
We further investigated the LSC/IRb/SSC/IRa borders in the chloroplast genomes of V. thibetica and eight Apiaceae, including C. paradoxum, B. chinense, L. sinense, A.sinensis, S. gyirongensis, P. praeruptorum, H. sibthorpioides, and H. yungningense (Fig. 7). As shown in Fig. 7, ycf1 was situated at the SSC/IRa boundary of all nine chloroplast genomes, indicating that it is a universal characteristic of the Apiaceae cp genome. At the IRb/SSC junction, ndhF and the duplicated pseudogene ycf1 had 3 and 37 bp overlapping in the V. thibetica and H. sibthorpioides cp genomes, and the duplicated ycf1 gene was already absent in C. paradoxum, B chinense, and S. gyirongensis. Except for V. thibetica and B. chinense, the other cp genomes contained trnH genes.

Comparison of the borders of the large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among nine Apiaceae chloroplast genomes. JLA, junction between LSC and IRa; JLB, junction between LSC and IRb; JSA, junction between SSC and IRa; JSB, junction between SSC and IRb
Furthermore, no novel genes were observed in the cp genome of V. thibetica compared with the gene composition of other Apiaceae family species, yet two genes (psbA and trnH) were lost. The trnL gene was only observed in C. paradoxum and V. thibetica. Finally, it was worth mentioning that the novel gene ORF8 was observed in the A. sinensis cp genome. The above results demonstrated that the plastid genomes of these nine Apiaceae species were relatively divergent.
Highly variable hotspot analysis of chloroplast genomes
Nucleotide diversity (Pi) values of plastomes among nine species were calculated using a sliding window to evaluate the level of diversity in different regions with species including V. thibetica, C. paradoxum, B. chinense, L. sinense, A. sinensis, S. gyirongensis, P. praeruptorum, H. sibthorpioides, and H. yungningense (Fig. 8). Highly divergent sites among nine Apiaceae species of cp genomes were detected using DNA polymorphism analysis. There were 17,234 variable sites, 3483 parsimony informative sites, and 1459 indels detected in nine plastid genomes, causing Pi values ranging from 0 to 0.1313. Meanwhile, we observed low variability in IRs compared to LSC and SSC partitions. Overall, the average nucleotide diversity within the nine Apiaceae cp genomes was 0.03650, representing a high divergence level among those species. Afterwards, it was further confirmed that the regions showing the higher Pi value peaks (> 0.09) were rps16 (pi = 0.11), ndhc-trnV-UAC (pi = 0.09), clpP (pi = 0.10), ycf1 (pi = 0.13), ndhB (pi = 0.11), including four gene regions and one intergenic region. These variable loci may provide valuable information for resolving species identification, phylogenetic relationships, and genetic diversity.

Analysis of sliding windows across chloroplast genomes in nine Apiaceae. X-axis: position of the window's midpoint; Y-axis: nucleotide diversity (Pi) per window. The red line represents the threshold value for variant loci (Pi threshold = 0.09). The high variation loci of the Apiaceae genome were marked in the figure. LSC, large single-copy; IRb, inverted repeat; SSC, small single-copy; IRa, inverted repeat
Phylogenetic analysis of the cp genomes
We constructed a maximum likelihood (ML) tree with 37 entire plastid genomes to determine the phylogenetic position of V. thibetica (Fig. 9). Of these, we downloaded 36 already published complete chloroplast genomes containing ten genera from NCBI (Table S5). The Eleutherococcus trifoliatus and Aralia cordata genome sequences of the Araliaceae family were used as outgroup. In parallel, entire topology of the ML tree and the node support for the principal clades were consistent with the NJ (Neighbor-Joining) tree (Fig. S1). As shown in Fig. 9, all species submitted for analysis were divided into three clades, corresponding to the Apioideae, Hydrocotyloideae subfamily, and outgroup, respectively. Here, V. thibetica was gathered into a single branch in the subfamily Apioideae, which verified the classification research of the previous scientists on the Apiaceae (Apioideae) (Watson 1998). Furthermore, V. thibetica was clustered together with genera Angelica, Peucedanum, and Ligusticum. A close relationship among them was also uncovered. Overall, Vicatia should be considered a separate genus in the Apiaceae family.

Plastome-based phylogenetic relationship among 37 Apiaceae species. The phylogenetic tree was constructed with the maximum likelihood (ML) method. The plastome sequences that E. trifoliatus and A. cordata of Araliaceae were used as outgroup. Values beside branch nodes denote support values for the bootstrap
Discussion
Genomic comparison
The chloroplast genome of V. thibetica reported in this study exhibited a circular quadripartite structure and was 145,796 bp in length, similar to the sequence of L. sinensis (Table 3). Thus, it indicated that V. thibetica and L. sinense were related species compared with other Apiaceae species. However, we have also noted that the cp genomes of the nine Apiaceae species, including V. thibetica, were varied in length (145,796–155,545) (Table3) (Wu et al. 2020; Li et al. 2019; Zheng et al. 2019, 2020; Zhang et al. 2019a, b; Tian et al. 2019; Xiao et al. 2019; Ge et al. 2017). Generally, the expanded IR regions and variable SSC regions were considered the main factors which were contributing to the chloroplast genome length variation in angiosperms (Kim and Lee 2004).
Furthermore, the difference was relatively significant in the comparative analysis of V. thibetica and A. sinense. The novel gene ORF8 was observed in the chloroplast genome of A. sinensis, but not in the V. thibetica. Insertions and deletions of genes were observed in the cp genomes of V. thibetica and A. sinense, which could be molecular markers to identify the two species (Fig. 7). At the same time, the variable fragments obtained from the whole genome alignment of V. thibetica and A. sinense also provided important information for their plant identification (Fig. 6). Despite their similarity in efficacy and morphology, they can be distinguished based on chloroplast genome super-barcode.
Variation of apiaceae cp genome
A common feature of Apiaceae chloroplast genomes revealed by genomic variation analysis is that the regions of IR were more conservative compared to LSC and SSC. Moreover, the variation level was significantly greater in non-coding compared to coding regions. In general, non-coding regions could be divided into intronic and intergenic regions, whereas intergenic spacers were highly variable among Apiaceae species in this study (Shaw et al. 2007). Previously, highly variable hotspots in the cp genome have been demonstrated to be useful for species identification and analysis of phylogeny while providing critical information at the population level to explore species differences and population structure (Bi et al. 2018; Du et al. 2017). Therefore, the rps16, ndhc-trnV-UAC, clpP, ycf1, and ndhB variable hotspots obtained by sliding window analysis (Fig. 8) could be used not only as molecular markers for the identification of V. thibetica and A. sinense, but also for the phylogenetic investigation of Apiaceae species. Similar results associated with these variant regions were also reported in recent researches of Fritillaria (Bi et al. 2018; Chen et al. 2019) and Dioscoreales (Biju et al. 2019).
Repeat analysis
Thirty-seven long repeats of 30–59 bp were identified in the cp genome of V. thibetica. However, in the reported species of the genus Ligusticum, 308 repeats of 30–82 bp were identified (Ren et al. 2020), indicating that long repeats are variable between related species and may serve as molecular markers for species identification (Nie et al. 2012). We observed highest number of SSRs in the LSC region, and the reasonable explanation is that the LSC region is more extended compared to SSC and IR (Ren et al. 2020). Among these SSRs, the A/T units of mononucleotide repeats are the most abundant, which may be explained by the higher proportion of polyA and polyT in the cp genome (Zhu et al. 2020). The identified SSRs may provide candidate SSR markers for molecular genetic correlation studies of medicinal plants in the genus Vicatia.
Phylogenetic analysis
We reconstructed ML and NJ trees using the whole cp genomes of 37 species, including V. thibetica. First, the Apioideae and Hydrocotyloideae subfamilies were clustered separately in two large clades of the phylogenetic tree, and these two large clades were further divided into distinct clades. Then, Vicatia, Angelica, Peucedanum, Semenovia, Ligusticum, Heracleum, Bupleurum, and Chamaesium clustered in Apioideae. And then, Angelica, Vicatia, Peucedanum, Ligusticum clustered in one smaller branch. On this smaller branch, when assigned, the genera Angelica, Peucedanum, and Ligusticum are sister groups, whereas Vicatia forms a separate branch independently. These observations indicated that the genera Vicatia, Angelica, Peucedanum, Ligusticum have been closely related, whereas V. thibetica was retained in the genus Vicatia. Our results have been consistent with previous studies (Pu 2005). However, the phylogenetic position of V. thibetica is still not sufficiently clear due to the lack of chloroplast genomic information for other Vicatia species. Therefore, it is indispensable to increase the cp genome of other Vicatia species.
Conclusions
This study reported the first chloroplast genome of V. thibetica and compared it with other Apiaceae species. The chloroplast genome of V. thibetica was similar in size, number of genes, genomic structure, and gene order to other angiosperm chloroplast genomes. We detected 75 SSRs, 29 tandem repeats, and 37 long repeats useful for genetic breeding and population genetics studies within Vicatia. In addition, the highly variable sites and divergent regions of nine Apiaceae species were identified as possible pathways for further use in studying genetic markers for population genetics. Comparative analysis with other species of the family Apiaceae showed that the IR regions were more conservative compared to the SSC and LSC, suggesting that more DNA barcodes could be developed from these regions to identify species. Meanwhile, the chloroplast genome could be used to distinguish V. thibetica from A. sinensis. Therefore, it may be unreasonable to use V. thibetica instead of traditional Chinese medicine A. sinensis. The phylogenetic study based on 37 complete chloroplast genomes showed that V. thibetica formed a single clade within the Apioideae subfamily, supporting the earlier view that Vicatia is an independent genus of the family Apiaceae (Watson 1998; Pu 2005).
Data availability
The data that support the findings of this study are publicly available in the GenBank of the NCBI database under Accession Number MZ189732.
Abbreviations
- Cp:
-
Chloroplast
- IR:
-
Inverted repeat
- LSC:
-
Large single-copy
- SSC:
-
Small single-copy
- PCGs:
-
Protein-coding genes
- rRNAs:
-
Ribosomal RNA genes
- tRNAs:
-
Transfer RNA genes
- SSR:
-
Simple sequence repeat
- RSCU:
-
Relative synonymous codon usage
- ML:
-
Maximum likelihood
- NJ:
-
Neighbor-joining
- CDS:
-
Protein-coding regions
- IGS:
-
Intergenic spacer
References
Amiryousefi A, Hyvönen J, Poczai P (2018) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34:3030–3031. https://doi.org/10.1093/bioinformatics/bty220/4961430
Asaf S, Khan AL, Khan MA (2018) Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species. PLoS ONE 13:e0192966. https://doi.org/10.1371/journal.pone.0192966
Bausher MG, Singh ND, Lee SB, Jansen RK, Daniell H (2006) The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var “Ridge Pineapple”: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol 6:21. https://doi.org/10.1186/1471-2229-6-21
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580. https://doi.org/10.1093/nar/27.2.573
Bi Y, Zhang MF, Xue J, Dong R, Du YP, Zhang X (2018) Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria. Sci Rep 8:397–416. https://doi.org/10.1038/s41598-018-19591-9
Biju VC, Shidhi PR, Vijayan S, Rajan VS, Sasi A, Janardhanan A, Nair AS (2019) The Complete chloroplast genome of Trichopus zeylanicus, and phylogenetic analysis with dioscoreales. Plant Genome 12(3):1–11. https://doi.org/10.3835/plantgenome2019.04.0032
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Brudno M, Malde S, Poliakov A, Do CB, Couronne O, Dubchak I, Batzoglou S (2003) Glocal alignment: finding rearrangements during alignment. Bioinformatics 19(Suppl 1):i54–i62. https://doi.org/10.1093/bioinformatics/btg1005
Chen Q, Wu XB, Zhang DQ (2019) Phylogenetic analysis of Fritillaria cirrhosa D. Don and its closely related species based on complete chloroplast genomes. Peer J 7:7e7480. https://doi.org/10.7717/peerj.7480
Dierckxsens N, Mardulyn P, Smits G (2016) NOVOPlasty:de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18. https://doi.org/10.1093/nar/gkw955
Dong WP, Liu J, Yu J, Wang L, Zhou SL (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7(4):e35071. https://doi.org/10.1371/journal.pone.0035071
Dong WP, Liu H, Xu C, Zuo YJ, Chen ZJ, Zhou SL (2014) A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet 15:138. https://doi.org/10.1186/s12863-014-0138-z
Dong ST, Zhang XQ, Hu Y (2018) General situation of chemical composition, quality control and pharmacology of Xigui. Chin J Ethnomed Ethnopharm 27(13):40–42
Du Y, Bi Y, Yang F, Zhang M, Chen X, Xue J, Zhang X (2017) Complete chloroplast genome sequences of Lilium: insights into evolutionary dynamics and phylogenetic analyses. Sci Rep 7:233–252. https://doi.org/10.1038/s41598-017-06210-2
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273–W279. https://doi.org/10.1093/nar/gkh458
Ge L, Shen LQ, Chen QY, Li XM, Zhang L (2017) The complete chloroplast genome sequence of Hydrocotyle sibthorpioides (Apiales: araliaceae). Mitochondrial DNA Part B 2(1):29–30. https://doi.org/10.1080/23802359.2016.1241676
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41(41):95–98. https://doi.org/10.1021/bk-1999-0734.ch008
Jansen RK, Cai ZQ, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J (2007) Analysis of 81genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci 104:19369–19374. https://doi.org/10.1073/pnas
Jiang B, Zhao G, Zhang DQ (2016) An illustrated book of bai nationality medicinal plants. Chinese Medicine Press, Beijing, p 156
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30(4):772–780. https://doi.org/10.1093/molbev/mst010
Kim KJ, Lee HL (2004) Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res 11(4):247–261. https://doi.org/10.1093/dnares/11.4.247
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29(22):4633–4642. https://doi.org/10.1093/nar/29.22.4633
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29(6):1695–1701. https://doi.org/10.1093/molbev/mss020
Li YS, Geng ML, Xu ZL, Wang Q, Li LL, Xu M, Li MM (2019) The complete plastome of Peucedanum praeruptorum (Apiaceae). Mitochondrial DNA Part B 4(2):3612–3613. https://doi.org/10.1080/23802359.2019.1676180
Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52(5–6):267–274. https://doi.org/10.1007/s00294-007-0161-y
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30(5):1188–1195. https://doi.org/10.1093/molbev/mst024
Nguyen L, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274. https://doi.org/10.1093/molbev/msu300
Nie XJ, Lv SZ, Zhang YX, Du XH, Wang L, Biradar SS, Tan XF, Wan FH, Weining S, Kolokotronis S (2012) Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 7(5):e36869. https://doi.org/10.1371/journal.pone.0036869
Park I, Yang S, Kim W, Song JH, Lee HS, Lee HO, Lee JH, Ahn SN, Moon BC (2019) Sequencing and comparative analysis of the chloroplast genome of Angelica polymorpha and the development of a novel indel marker for species identification. Molecules 24(6):1038. https://doi.org/10.3390/molecules24061038
Powell W, Morgante M, Andre C, McNicol JW, Machray GC, Doyle JJ, Tingey SV, Rafalski JA (1995) Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr Biol 5(9):1023–1029. https://doi.org/10.1016/S0960-9822(95)00206-5
Pu FD, Mark FW (2005) Vicatia DC. In: Wu ZY, Hong DY, Raven PH (eds) Flora of China, vol 14. Science Press and Missouri Botanical Garden Press, Beijing and St Louis, pp 52
Pu FD (2005) Taxonomic notes on Meeboldia H. Wolff (Umbelliferae). Acta Phytotaxonomica Sinica 43(6):552. https://doi.org/10.1360/aps030076
Raven JA, Allen JF (2003) Genomics and chloroplast evolution: What did cyanobacteria do for plants? Genome Biol 4(3):209. https://doi.org/10.1186/gb-2003-4-3-209
Ren T, Li ZX, Xie DF, Gui LJ, Peng C, Wen J, He XJ (2020) Plastomes of eight Ligusticum species: characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol 20(1):519–519. https://doi.org/10.1186/s12870-020-02696-7
Roullier C, Rossel G, Tay D, McKey D, Lebot V (2011) Combining chloroplast and nuclear microsatellites to investigate origin and dispersal of New World sweet potato landraces. Mol Ecol 20(19):3963–3977. https://doi.org/10.1111/j.1365-294X.2011.05229.x
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Sharp PM, Tuohy Therese MF, Mosurski Krzysztof R (1986) Codon usage in yeast: cluster analysis clearly diferentiates highly and lowly expressed genes. Nucleic Acids Res 14:5125–5143. https://doi.org/10.1093/nar/14.13.5125
Shaw J, Lickey EB, Schilling EE, Small RL (2007) Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot 94:275–288
She ML, Pu FD, Pan ZH, Mark FW (1979) Apiaceae lindley. In: Wu ZY, Hong DY, Raven PH (eds) Flora of China, vol 55. Science Press and Missouri Botanical Garden Press, Beijing and St Louis, pp 185
Tang P, Ruan QY, Peng C (2011) Phylogeny in structure alterations of poaceae cpDNA. Chin Agric Sci Bull 27:171–176
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106(3):411–422. https://doi.org/10.1007/s00122-002-1031-0
Tian EW, Liu QQ, Chen WN, Li F, Chen AM, Li C, Chao Z (2019) Characterization of complete chloroplast genome of Angelica sinensis (Apiaceae), an endemic medical plant to China. Mitochondrial DNA Part B 4(1):158–159. https://doi.org/10.1080/23802359.2018.1544862
Watson MF (1998) Notes relating to the flora of Bhutan: XXXVI. Umbelliferae, II. Edinburgh J Bot 55(3):367–415. https://doi.org/10.1017/S0960428600003267
Wu QW, Wu H, Wang LK, Zhao X (2020) Characterization of the complete chloroplast genome of Ligusticum sinense, as a Chinese herb to treat toothache in China. Mitochondrial DNA Part B 5(3):3174–3175. https://doi.org/10.1080/23802359.2020.1808103
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17):3252–3255. https://doi.org/10.1093/bioinformatics/bth352
Xiao QY, Feng T, Yu Y, Luo Q, He XJ (2019) The complete chloroplast genome of Semenovia gyirongensis (Tribe Tordylieae, Apiaceae). Mitochondrial DNA Part B 4(1):1863–1864. https://doi.org/10.1080/23802359.2019.1613199
Zhang W, Duan ZH, Sun F (2004) The Chemical constituents from the root of Vicatia Thibetica. Nat Prod Res Dev 16:218–219
Zhang ZL, Zhang Y, Song MF, Guan YH, Ma XJ (2019a) Species identification of Dracaena using the complete chloroplast genome as a super-barcode. Front Pharmacol 10:1441. https://doi.org/10.3389/fphar.2019.01441
Zhang F, Zhao ZY, Yuan QJ, Chen SQ, Huang LQ (2019b) The complete chloroplast genome sequence of Bupleurum chinense DC. (Apiaceae). Mitochondrial DNA Part B 4(2):3665–3666. https://doi.org/10.1080/23802359.2019.1678427
Zheng HY, Guo XL, He XJ, Yu Y, Zhou SD (2019) The complete chloroplast genome of Chamaesium paradoxum. Mitochondrial DNA Part B 4(1):2069–2070. https://doi.org/10.1080/23802359.2019.1617064
Zheng ZY, Li J, Xie DF, Zhou SD, He XJ (2020) The complete chloroplast genome sequence of Heracleum yungningense. Mitochondrial DNA Part B 5:1783–1784. https://doi.org/10.1080/23802359.2020.1749150
Zhou N, Duan YM, Chen Q, Ma XK (2007) Study on Pharmacognosy of Xigui. J Anhui Agric Sci 35(8):2307–2425. https://doi.org/10.1398/j.cnki.0517-6611.2007.08.059
Zhu B, Feng Q, Yu J, Yu Y, Zhu XX, Wang Y, Guo J, Hu X, Cai MX (2020) Chloroplast genome features of an important medicinal and edible plant: Houttuynia cordata (Saururaceae). PLoS ONE 15(9):e239823. https://doi.org/10.1371/journal.pone.0239823
Acknowledgements
We thank Dr. Jun Qian of Biozeron Biotech Co. Ltd., Shanghai, China, for his assistance in data analysis and the anonymous reviewers for helpful comments and valuable views on the manuscript.
Funding
This work was supported by the Major Projects of Science and Technology Plan of Dali state (D2019NA03) and Li Jian Expert Workstation of Yunnan Province (202005AF150013).
Author information
Authors and Affiliations
Contributions
Conceptualization, YG and CX; methodology, CX and BD; software, YG and WL; investigation, YG and CX; resources, YG; CX; BD; HZ; XC; YW; writing—original draft preparation, YG; writing—review and editing, CX and BD; supervision, CX; project administration, CX; BD; HZ; XC; YW; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guan, Yh., Liu, Ww., Duan, Bz. et al. The first complete chloroplast genome of Vicatia thibetica de Boiss.: genome features, comparative analysis, and phylogenetic relationships. Physiol Mol Biol Plants 28, 439–454 (2022). https://doi.org/10.1007/s12298-022-01154-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-022-01154-y