
Abstract
In this work O-linked glycopeptides bearing mucin core-1 type structures were enriched from human serum. Since about 70 % of the O-glycans in human serum bind to the plant lectin Jacalin, we tested a previously successful protocol that combined Jacalin affinity enrichment on the protein- and peptide-level with ERLIC chromatography as a further enrichment step in between, to eliminate the high background of unmodified peptides. In parallel, we developed a simpler and significantly faster new workflow that used two lectins sequentially: wheat germ agglutinin and then Jacalin. The first lectin provides general glycopeptide enrichment, while the second specifically enriches O-linked glycopeptides with Galβ1-3GalNAcα structures. Mass spectrometric analysis of enriched samples showed that the new sample preparation method is more selective and sensitive than the former. Altogether, 52 unique glycosylation sites in 20 proteins were identified in this study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein glycosylation is among the most prevalent post-translational modifications (PTM). Glycosylation has been implicated in several physiological and pathological events, including proper folding and half-life of proteins [1], protein-protein interactions, cell-cell communication [2, 3] and disease [4–12]. In order to understand the exact role of glycans in these events, detailed characterization of glycoproteins is required. For biomarker studies, readily available body fluids including serum are the preferred sample sources.
For large scale PTM studies efficient enrichment strategies, as well as good mass spectrometric characterization methods, are required. O-glycopeptides represent a significant challenge from both aspects. The huge background of abundant unmodified proteins and those only N-glycosylated hinders efficient O-glycosylation profiling of body fluids. In addition, glycosylation displays heterogeneity both in site occupancy and in the number of structures at any given site. Different enrichment strategies have been tested for the selective isolation of O-glycopeptides. Hydrophilic interaction liquid chromatography (HILIC) showed limited success for O-glycopeptides [13] as this method is more amenable to the separation of glycopeptides displaying large glycans, such as N-glycopeptides. Another enrichment approach, restricted to sialic acid-containing glycoproteins, is the periodate oxidation-hydrazide capture strategy [14, 15], which has been applied for selective isolation of glycopeptides from tryptic digests of human urine [16] and cerebrospinal fluid [17]. Finally, the specific interactions of glycans with lectins can also be exploited. Vicia villosa agglutinin has been used to isolate O-glycopeptides from bioengineered cell lines expressing exclusively Tn or sialo Tn antigens [18–22]. Wheat germ agglutinin has been applied for simultaneous isolation of N- and O-glycopeptides from the mouse synaptosome and liver [23, 24]. Finally, Jacalin has been used for the isolation of mucin core-1 type O-glycopeptides from fetal calf serum [25, 26].
Mass spectrometric characterization of O-glycopeptides is also challenging. Collisional activation usually results in MS/MS spectra dominated by glycosidic bond cleavages. Ion trap CID spectra feature abundant sugar losses from the precursor ions, some oxonium ions, but practically no peptide fragments. While beam-type CID (HCD) spectra display peptide fragments as well, the gas-phase elimination of the glycan(s) is usually complete [27]. As a result, site assignment from CID data is rarely possible. On the other hand, with electron-capture dissociation (ECD) [28] and electron-transfer dissociation (ETD) [29] the peptide backbone is fragmented while the glycans are mostly unaffected. ETD is more efficient than ECD, but the quality of the resulting spectra depends on the charge-density of the precursor ion [30]. As the modifying glycans increase the mass of the peptide, usually without adding extra charge, glycopeptides generally have a low charge-density. Removing sugar units with sequential exoglycosidase digestions increases the charge-density [25], although information about the original glycan structure and heterogeneity is evidently lost. Clustered potential modification sites, multiple glycosylations on the same peptide and Pro residues close to the modified residue (Xxx-Pro bonds are not cleaved under ECD/ETD conditions), all common occurrences in O-glycosylation, make site assignments harder. However, despite the limitations of the ECD/ETD activation, all O-glycosylation studies on complex protein samples reported to date have been performed using these techniques [16–26].
Software interpretation of glycopeptide spectra presents many challenges. More than 20 GalNAc transferases may initiate O-glycosylation [31], and no universal consensus motif exists. In addition, tyrosine glycosylation has been reported recently [32]. Thus, all Ser, Thr and Tyr residues have to be considered as potential candidates in database searches. The number of potential glycan structures opens up the search space even further. One can specify the potential oligosaccharide structures for the database search by relying on existing glycan databases, by analyzing the released glycans to create a library of compositions to consider, or by exploring the glycan pool by performing iterative database searches with undefined modifications at potential glycosylation sites [23, 24]. We are familiar with two search engines, Protein Prospector and Byonic that are equally suitable for the analysis of ETD data of O-linked glycopeptides [27, 33]. When only beam-type CID data are available, gas-phase elimination of the sugar units has to be permitted, and assigning oxonium ions and reducing terminal Y-type glycosidic fragments [34] is also important. Byonic usually handles such data better than Protein Prospector, especially when the modifying glycans are oligosaccharides, not just single HexNAc units. Unfortunately, it always assigns a modification site, even when there is no evidence for locating the site. Protein Prospector has a built-in site localization in peptide (SLIP) score and indicates the glycosylation site, by default, only when its probability is at least 95 %; otherwise, all potential sites are listed [35]. Database searches with HCD data permitting oligosaccharides have to be ‘customized’ for this search engine, as described in the “Experimental” section, and the oxonium ions are not assigned.
In the present study we wanted to enrich O-linked glycopeptides from human serum without having to perform an extra step to deplete it of the most abundant proteins. We aimed at the isolation of glycopeptides modified with mucin-type core-1 structures, as this glycoform has been reported to be the most abundant in human serum [36]: NeuAcα2-3Galβ1-3GalNAcα (~60 %), NeuAcα2-3Galβ1-3(NeuAcβ2-6)GalNAcα (~20 %), and Galβ1-3GalNAcα (~10 %) represent ~90 % of the total O-glycan pool [37]. Considering the wide dynamic range of serum protein levels, and the expected heterogeneity of O-glycosylation, we followed multistep enrichment strategies. We reasoned that protein-level affinity enrichment using Jacalin, which preferentially binds the Galβ1-3GalNAcα structure, would remove most of the unmodified or only N-glycosylated proteins. Previously, we found that peptide-level enrichment using Jacalin from the tryptic digest of Jacalin-bound protein mixtures yielded glycopeptides with a very high non-specific background [25]. Thus, we decided to add a further fractionation method in between these two steps that operates using both hydrophilicity and acidity: electrostatic repulsion hydrophilic interaction chromatography (ERLIC [38]), which at least partially separates glycopeptides from unmodified peptides. We also employed exoglycosidase treatment to improve the charge-density of the glycopeptides by retaining only the core GalNAc(s). The sample preparation outlined above permitted efficient glycopeptide enrichment from fetal calf serum [26]. This protocol yields decent results, but is labor and time intensive. In search of a faster and more efficient workflow, we developed a new enrichment strategy. A few years ago we discovered that another lectin, wheat germ agglutinin (WGA), shows a weak affinity towards glycopeptides in general and the enrichment is quite efficient [23]. Thus, we first performed a glycopeptide enrichment from human serum tryptic digest using WGA. Then, to target the O-linked glycopeptides, we used lectin affinity chromatography with Jacalin. Prior to the second enrichment step a neuraminidase digestion was performed, hoping that previously missed disialo structures (representing about 20 % of the glycan pool) would also be isolated once the sialic acid from the core GalNAcα was removed.
We compared the former enrichment protocol with the new method. In both workflows, glycopeptides were analyzed by HCD and ETD activation. For data interpretation we used two different search engines, Protein Prospector and Byonic, and carefully inspected the assignments. Altogether we identified 52 glycosylation sites in 20 proteins in this study.
Experimental
Human serum was from Sigma (H4522). Bovine trypsin (Sigma T1426) was used to digest proteins.
Sample preparation 1 (SP1)
One milliliter of human serum was injected onto a 4.6 × 50 mm column (column volume (CV): 830 μl) packed with agarose-bound Jacalin (Vectorlabs AL1153). After loading the sample (flow rate: 200 μl/min), the column was washed with ~40 CV of solvent A (175 mM Tris(hydroxymethyl)aminomethane (Tris), pH: 7.5, flow rate: 1 ml/min), then the bound proteins were eluted by injecting a 2 ml plug of 0.8 M galactose in solvent A (flow rate: 200 μl/min). Collected glycoprotein fractions were concentrated using 10 kDa MWCO centrifugal filter units (Amicon Ultra-15, Millipore), then digested with trypsin according to the FASP protocol [39]. Tryptic peptides were then subjected to further glycopeptide enrichment steps.
ERLIC chromatography was performed on a weak anion exchange column (PolyWAX LP, PolyLC Inc, 4.6 × 200 mm, 5 μm particle size, 300 Å pore size) applying the following gradient program: 0–5 min: 0 % B, 5–15 min: 0–10 % B, 15–35 min: 10–60 % B, 35–45 min: 60–100 % B, 45–65 min: 100 % B (flow rate: 1 ml/min, UV-detection at 215 nm). The following solvents were used: ERLIC1 (E1): solvent A: 20 mM methyl-phosphonic acid pH: 2 / 70 % acetonitrile (ACN), solvent B: 200 mM triethylammonium phosphate (TEAP) pH: 2 / 60 % ACN (the pH of solvent A and B were adjusted using 10 M aqueous NaOH and triethylamine, respectively); ERLIC2 (E2): solvent A: 20 mM formic acid / 70 % ACN, solvent B: 1 M formic acid / 10 % ACN. Two-minute fractions were collected between 0 and 60 min (altogether 30 fractions), and dried down. E1 samples were desalted on C-18 tips (Omix, Varian) and concentrated.
Each fraction was subjected to glycopeptide enrichment using a 1 × 100 mm column (CV: 78.5 μl) packed with agarose-bound Jacalin (Vectorlabs AL1153). After introducing the sample (flow rate: 50 μl/min), the column was washed with ~50 CV of solvent A (175 mM Tris, pH: 7.5, flow rate: 150 μl/min), then the bound peptides were eluted by injecting a 500 μl plug of 0.8 M galactose in solvent A (flow rate: 150 μl/min). Collected glycopeptide fractions were concentrated and desalted using C18 tips (Omix, Varian). The resulting glycopeptide mixtures were subjected to exoglycosidase treatment using α2-3,6,8 neuraminidase (5 h at 37 °C; 2.5 U/sample, New England Biolabs P0720; in 50 mM sodium citrate, pH: 6.0) followed by treatment with β1−3 galactosidase (overnight at 37 °C; 2.5 U/sample, New England Biolabs P0726; in 100 mM sodium citrate, pH: 4.5). Deglycosylation was stopped by acidification to pH ≤ 3 with 10 % TFA solution, and the resulting peptide mixtures were desalted on C18 tips (Millipore ZTC18S960).
Sample preparation 2 (SP2)
Four hundred microliter human serum was digested with trypsin according to the FASP protocol [39] and enriched in 5 separate injections on a 2 × 250 mm column (CV: 785 μl) packed with WGA immobilized on POROS Al resin [40] as follows. After loading the sample, the column was washed with WGA buffer (~2 CV; 100 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 5 % ACN; flow rate: 125 μl/min), then a 100 μl plug of 200 mM N-acetyl-D-glucosamine in WGA buffer was injected at 12 min. Three-minute fractions were collected between 0 and 24 min. Fractions 4–8 were acidified, desalted using C18 tips (Omix, Varian), combined and dried down before treating with α2-3,6,8 neuraminidase (100 U, New England Biolabs P0720; in 50 mM sodium citrate, pH: 6.0 for 7.5 h at 37 °C). The pH of the deglycosylation mixture was set to 7.5 using 1.75 M Tris prior to glycopeptide enrichment using Jacalin (see peptide-level enrichment described for SP1).
Mass spectrometry
The isolated glycopeptide mixtures were analyzed by LC-MS/MS using a Waters nanoAcquity UPLC on-line coupled to a linear ion trap-Orbitrap (Orbitrap Elite, Thermo Fisher Scientific) mass spectrometer operating in positive ion mode. Thirty-three percent of the isolated peptide mixtures were injected for each LC-MS/MS analyses. After trapping at 3 % B (Waters Symmetry C18 180 μm × 20 mm column, 5 μm particle size, 100 Å pore size; flow rate: 10 μl/min), peptides were separated using a linear gradient of 10 to 40 % B in 30 or 90 min for SP1 or SP2, respectively (using a 75 μm × 90 mm column self-packed with MagicC18AQ, 3 μm particle size, 200 Å pore size; solvent A: 0.1 % formic acid/water, solvent B: 0.1 % formic acid/ACN; flow rate: 400 nl/min). Data acquisition was carried out in a data-dependent fashion, the 3 most abundant, multiply charged ions were selected from each MS survey scan (m/z: 380–1400) for MS/MS analyses. HCD data were acquired for each precursor, while ETD data acquisition was triggered by the presence of diagnostic sugar oxonium ions m/z 204.0867 (for N-acetylhexosamine, HexNAc) or 366.1395 (for hexosyl N-acetylhexosamine, HexNAcHex) among the 50 most abundant fragments of the HCD spectrum, the mass accuracy requirement was 15 ppm. MS and HCD spectra were acquired in the Orbitrap, and ETD spectra in the linear ion trap. Supplemental activation for the ETD experiments was enabled (supplemental activation energy: 15). Normalized collision energy (NCE) for HCD experiments was set to 35. Dynamic exclusion was enabled (exclusion time: 60 s). For SP2, MS/MS data of precursors with z = 2 and z > 2 charge states were acquired in 2 consecutive experiments.
Data interpretation
Proteome Discoverer (Thermo Scientific, v1.4.0.288) was used to generate separate HCD and ETD peak lists from the raw data. HCD peak lists were filtered for potential glycopeptide data using the MS-Filter program of Protein Prospector [41]: only those spectra that contained the characteristic HexNAc oxonium ion m/z: 204.0867 ± 10 ppm within the 40 most abundant fragment ions were retained. HCD spectra not featuring the HexNAc oxonium ion were also saved in a separate peak list for identifying the unmodified background.
The MS-Filter program was also used for estimation of enrichment and exoglycosidase treatment efficiency or for finding spectra containing other glycan-specific fragment ions. In all cases, required m/z values were searched within 10 ppm mass accuracy in the 40 most abundant fragment ions.
All peak lists were searched using Protein Prospector (v5.12.3.) with mass accuracies of 5 ppm for precursor ions and 10 ppm (for HCD) or 0.6 Da (for ETD) for fragment ions specified as monoisotopic values. Additional search parameters are listed in Table 1. In order to identify multiply modified peptides from HCD data, respective database searches were repeated on the subset of confidently identified glycoproteins with the following user defined modifications: 203.0794 (HexNAc), 406.1588 (2xHexNAc), and 609.2382 (3xHexNAc) for SP1; and 365.1322 (HexHexNAc), 730.2644 (2xHexHexNAc), and 1095.3966 (3xHexHexNAc) for SP2; all defined as neutral loss. ETD data were also searched for Tyr-glycosylation in separate searches using the same parameters described above except that the appropriate glycan structures were allowed on Ser, Thr and Tyr residues and only one Tyr modification was allowed per peptide.
In order to evaluate the non-specific background, HCD data (using the 204-depleted peak lists) were also searched in the human subset of the Uniprot database (06.11.2014 version, 136244 sequences) applying the same search parameters as described above except that semitryptic peptides with maximum 1 missed tryptic cleavage site were considered, glycan modifications were not allowed, and maximum 2 variable modifications per peptide were permitted.
Acceptance criteria for all searches were: minimum scores: 22 and 15; maximum E values: 0.01 and 0.05 for protein and peptide identifications, respectively; SLIP score: >6 [35]. Glycopeptide identifications meeting the acceptance criteria were also evaluated manually.
HCD data were also searched using the Byonic software (v2.0-25, Protein Metrics Inc.) with the following parameter set: human subset of the Uniprot database (downloaded 11/13/2013, 88500 entries); tryptic peptides with maximum one missed cleavage; mass accuracies within 5 ppm for precursor ions and 10 ppm for fragment ions; carbamidomethylation of Cys residues was set as a fixed modification; variable modifications considered were Met oxidation (common, maximum 3 modifications/peptide); cyclization of N-terminal Gln residues (rare, maximum 1 modification/peptide); protein N-terminal acetylation (rare, maximum 1 modification/peptide) and glycans corresponding to sample preparation: HexNAc on Ser/Thr (common, maximum 1 modification/peptide) for SP1, and HexNAcHex on Ser/Thr (common, maximum 1 modification/peptide) for SP2. Additionally, the “6 most common” O-glycans (containing HexNAc, HexNAc2, HexNAcHex, HexNAc2Hex, HexNAcHexSA, and HexNAcHexSA2) on Ser/Thr were also specified as rare variable modification for all searches. A total of 3 and 1 modifications per peptide were allowed for common and rare modifications, respectively. Reverse sequences were concatenated to forward entries and common contaminants (using the built-in “common contaminant” database of 69 proteins) were also considered. The acceptance criteria were a 2 % false discovery rate on the protein level and a minimum peptide score of 200.
Results
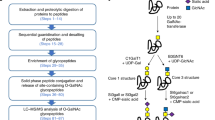
Two different sample preparation workflows were tested for the isolation and characterization of O-glycopeptides from human serum (Fig. 1). In the first set of experiments (SP1), we followed a workflow used successfully in O-glycosylation analysis of bovine serum [26]. A protein-level affinity-based enrichment using the lectin Jacalin, specific for Galβ1-3GalNAcα structure was performed. The tryptic digest of this enriched fraction was then separated by an acidity/hydrophilicity-based fractionation (ERLIC chromatography [38]), before fractions were subjected to a final Jacalin enrichment step. A new, volatile solvent system was also tested for the ERLIC fractionation of the glycoprotein digest. The isolated glycopeptides were treated with exoglycosidases to remove the sialic acid and galactose residues. This increases the charge-density of precursors, promoting more efficient ETD fragmentation [25]. In the SP2 workflow, glycopeptides were isolated from a human serum tryptic digest by sequential lectin affinity chromatography, first using wheat germ agglutinin (WGA) for general glycopeptide enrichment, followed by Jacalin for capturing O-glycopeptides. In addition, the WGA eluant was treated with neuraminidase to eliminate interference by sialic acids modifying the 6-OH of the core GalNAcα (disialo structures represent ~20 % of the O-glycan pool [37]). Isolated glycopeptide mixtures were analyzed by data-dependent LC-MS/MS. HCD data were acquired for all selected precursors, while ETD data were acquired only on glycopeptides, using the HCD fragment ion dependent ETD data acquisition [42]. HCD data were interrogated using the Protein Prospector and Byonic search engines, while ETD data were analyzed with Protein Prospector only. Results reported are based on Protein Prospector identifications and correspond to ~300 μl serum equivalent (based on the sample amount used for LC-MS/MS).

Sample preparation methods for O-glycopeptide enrichment
In the SP1 workflow, the protein-level affinity enrichment using Jacalin efficiently reduced non-specific background from unmodified or only N-glycosylated proteins as shown by the lower number of identified peptides representing abundant serum proteins such as serum albumin and serotransferrin in SP1 as compared to SP2 (Online Resource 1). In the subsequent ERLIC fractionation, in parallel with using the original sodium methyl-phosphonate/TEAP buffer (E1 [38]), we also tested a volatile solvent system utilizing formic acid (E2). The volatile formic acid/acetonitrile/water solvent system (E2) was found to be compatible with O-glycopeptide isolation, and this mass spectrometry-friendly solution delivered results identifying a higher number of glycosylation sites: 26 sites were identified with E2 as opposed to 18 sites with E1 (Table 2, Online Resource 1). The β1-3 galactosidase treatment was not entirely successful, approximately one third of the HCD-based glycopeptide identifications represented peptides modified with the disaccharide, both for E1 and E2 (Byonic identifications, data not shown). All of these glycopeptides were also identified bearing only the core GalNAcα. Filtering the HCD peak lists for the diagnostic oxonium ion of the disaccharide, m/z: 366.1395 revealed that 44 % of all detected glycopeptides in SP1 retained the galactose (Table 3). Neuraminidase treatment was found to be fairly efficient, less than 2 % of the HCD spectra featured the fragment ion m/z 274.0921 characteristic to sialic acid (Table 3).
The results from these two protocols together led to fewer identifications than our previous bovine serum results [26]. Hence, we designed a new, faster and simpler workflow, SP2. First, the sample was loaded onto a wheat germ agglutinin column, which binds both N- and O-linked glycan structures [23, 24] to produce a glycopeptide fraction. This glycopeptide mixture was then treated with neuraminidase, such that when the sample was then loaded onto a Jacalin column that captured only O-glycopeptides, including those that were previously sialylated on the core GalNAcα. Learning from our experience with SP1, β1-3 galactosidase treatment was not performed after the enrichment. Since we did not fractionate the glycopeptide mixture further off-line, we analyzed this sample twice performing ‘gas-phase fractionation’, i.e., in the first LC/MS analysis only doubly charged precursor ions were selected for MS/MS analysis, while in the second run these ions were excluded from the precursor selection. Both analyses yielded glycopeptide identifications. Judging from the presence of the diagnostic m/z 204.0867 oxonium ion, 34.4 % of the doubly charged ions and 76.4 % of the higher charge states represented glycopeptides, respectively. This indicates superior selectivity in comparison to the other workflow (Table 3). As a consequence, considerably more (46) glycosylation sites were identified from this sample preparation (Table 2, Online Resource 1).
Altogether 20 glycoproteins with 52 O-glycosylation sites were identified (Table 2; identifications representing glycosylation sites can be viewed using MS-Viewer [43]; search keys: ix1tmqvo4h (ETD data) and c05zbo4k0k (HCD data); the list of the identified glycopeptides can be found in Online Resource 1), of which 32 sites could be assigned unambiguously. The majority of the identified glycoproteins represent abundant serum proteins. All identified proteins except proteoglycan-4 and fetuin-B are listed in the 150 most abundant plasma proteins [44]. We successfully identified new modification sites: 21 of the 52 glycosylation sites are not reported in the Uniprot database (Table 2).
The tested sample preparation workflows provided partly overlapping results: 21 sites were observed both in SP1 and SP2 (Table 2). SP2 yielded more unique glycosylation site identifications: 6 and 25 sites were identified only with SP1 and SP2, respectively (Table 2). Overlap of the unambiguously assigned glycosylation sites is shown in Fig. 2. All unambiguously assigned sites represent glycosylation on Ser/Thr; Tyr-glycosylation was not detected.

Overlapping of the glycosylation sites assigned unambiguously from the different workflows
Discussion
Glycopeptide enrichment
Two different lectin-based sample preparation workflows were tested for the isolation of O-glycopeptides from human serum. SP1 is a previously successful, but labor and time intensive workflow; SP2 is much more streamlined. Both sample preparations prominently rely on Jacalin, a plant lectin that preferentially binds Galβ1-3GalNAcα. Previously, we have applied this lectin for the characterization of O-glycopeptides from fetal calf serum [25, 26]. Our lectin selection can be further justified by the composition of the human serum O-glycan pool. The dominance of the monosialylated mucin core-1 O-glycan, NeuAcα2-3Galβ1-3GalNAcα in human serum O-glycoproteins has been demonstrated by two independent studies [36, 37]. This structure represents more than 60 % of the glycans, followed by the diasialo and asialo variants, at approximately 20 and 10 %, respectively [37].
Based on our previous experience [26], in the SP1 workflow we combined protein- and peptide-level Jacalin affinity chromatrography with electrostatic repulsion hydrophilic interaction chromatography (ERLIC) to minimize the non-glycosylated background. ERLIC with volatile solvents has been successfully used in different proteomic experiments for protein identification, phosphorylation analysis and N-glycosylation studies [45–50]. We demonstrated here that the volatile formic acid/acetonitrile/water solvent system is also suitable for O-glycopeptide analysis, and eliminating a desalting step is definitely beneficial for the process. However, the number of identified glycosylation sites was far below our expectations – workflows E1 and E2 together delivered only 27 glycosylation sites while E1 yielded 117 O-glycosylation sites from fetal calf serum in our previous study, albeit from 2 ml starting material [26]. Incomplete removal of Galβ1-3 from the glycans (described in the “Results” section) certainly contributed to the lower performance of method SP1. At the same time, this yield for human serum seems to be reproducible, since SP1 with the two different solvent systems delivered comparable results.
In protocol SP2, a two-step peptide-level enrichment protocol was followed, using wheat germ agglutinin and then Jacalin. We decided to use WGA because in our previous glycopeptide enrichment experiments we found that this lectin binds both N- and O-linked glycopeptides indiscriminately [23]. It has been reported earlier that WGA binds not only GlcNAc and sialic acid as previously believed, but other glycan structures as well [51]. This new approach proved to be more selective and sensitive. A further advantage was the highly reduced time requirement of both sample preparation and MS analyses. In SP1, ERLIC fractionation resulted in 30 chromatographic fractions, all of them had to be subjected to subsequent Jacalin enrichment, exoglycosidase treatment, and desalting prior to LC-MS/MS analysis. SP2 yielded a single glycopeptide fraction that was analyzed by ‘gas-phase fractionationʼ of the components, i.e., separate acquisition of MS/MS data from z = 2 and higher charge state precursors during two extended LC-MS/MS analyses. The glycopeptides were present predominantly in z > 2 charge states, only one site assignment (Thr-628 in kininogen-1) was obtained exclusively from a doubly charged precursor ion (Online Resource 1). The data shown here were obtained from roughly the same serum equivalent (~300 μl based on the sample amount subjected to LC-MS/MS). Thus, some of the differences are most likely due to higher sample losses in the multistep SP1 protocol.
Even though SP2 outperformed SP1, the number of identified O-glycosylation sites is rather low. N-glycopeptides are also captured by WGA, and this might be a limiting factor in overall sensitivity. Removal of N-glycans prior to WGA chromatography using, for example, PNGase F (not performed in the present workflows) may improve the efficiency of SP2. Incomplete removal of sialic acids α2-6 linked to the core GalNAcα would compromise enrichment efficiency, since modification of the core GalNAcα 6-OH prevents Jacalin binding [52]. Windwarder et al. has recently reported that the neuraminidase applied in the present study (P0720 from New England Biolabs) did not cleave efficiently α2-6 linked sialic acid [53]. Thus, using a neuraminidase of different specificity might improve our yields.
All glycosylation sites identified in the present study are Ser or Thr residues. Judging from other large scale O-glycopeptide studies [18–20, 23], Ser/Thr modification occurs much more frequently, and perhaps at a higher level, than the glycosylation of Tyr. Thus, perhaps it is not surprising that we did not identify any Tyr-glycosylation in this study.
Our results show partial overlap with studies conducted on other body fluids [16, 17], indicating that their glycoprotein repertoire may vary, or that the alternative enrichment strategies capture only subportions of the O-glycoproteome and further improvements in sample preparation are required for comprehensive glycopeptide profiling.
Automated glycopeptide identification
In order to identify the highest possible number of glycopeptides, we applied multiple sets of search parameters in database searches using Protein Prospector (Table 1). Careful investigation of the results revealed that consideration of non-tryptic cleavages is necessary – many glycosylation sites (Thr-151 in kininogen-1; Thr-24, Thr-29, and Ser-30 in hemopexin; Thr-691 in ITI H2; Ser-1242/Thr-1244 in complement C4; Thr-47, and Thr-48 in plasma protease C1 inhibitor) were identified exclusively from semi-tryptic peptides (Online Resource 1). Rampant proteolysis in commercially available serum was observed in our pilot experiments [25, 26]. Presuming that O-glycosylation in proximity of cleavage sites may hinder tryptic digestion, we conducted database searches allowing for up to 2 missed cleavages. The majority of the glycopeptides were identified from peptides with none or 1 missed tryptic cleavage site (Online Resource 1). However, glycosylation sites Thr-47 and Thr-48 in plasma protease C1 inhibitor were identified only from peptides featuring 2 missed tryptic cleavage sites. Finally, the glycopeptide representing Thr-434/Thr-436 in vitamin D-binding protein (D6RF35) was only identified from the Uniprot database. In summary, comprehensive glycopeptide identification calls for search parameters that open up the search space, leading to higher false discovery rates. In addition, for sensitivity reasons, ETD data are often acquired in the ion trap. Due to the resulting low mass accuracy, the same fragment ions may be assigned to multiple fragments, leading to identifications less confident than indicated by the search engine, and may also result in less reliable modification site assignments. Products of glycan fragmentation processes (some do occur in ETD [25, 26]) may also be mistakenly assigned as peptide fragments. On the other hand, low-abundance fragment ions not used by Protein Prospector (this search engine uses only the 20 most abundant fragment ions from each half of the MS/MS spectrum by default) may improve the confidence of identifications. Thus, manual validation of glycopeptide assignments is still highly advisable. Since HCD data contain valuable information about the glycan structures in form of glycan-specific oxonium ions, the MS-Filter program of Protein Prospector [41] proved to be a highly valuable tool in our study. We used it to distinguish between glycopeptide and non-glycopeptide spectra, and to estimate the enrichment and exoglycosidase treatment efficiency. This software may prove to be highly useful for other applications whenever a diagnostic fragment ion or characteristic neutral loss ion is generated. For example, sulfopeptides could be identified from the diagnostic SO3 loss from the precursor ion.
Conclusions
We have presented our first attempts aimed at the characterization of O-glycosylation in human serum. Based on these data we believe that wheat germ agglutinin-based lectin affinity chromatography may hold the key for the enrichment of O-glycopeptides, after extended PNGaseF digestion to eliminate the interfering N-glycosylated components. One or two repeated enrichment steps with the same lectin may minimize the non-modified background, and will eliminate the need for using Jacalin. Following such a protocol O-glycopeptides featuring less frequently occurring glycans should also be isolated. Additional off-line fractionation, such as high pH reversed phase chromatography, may reveal glycopeptides representing less abundant proteins. Improved instrumentation is also available for glycopeptide analysis, the Orbitrap Fusion Lumos Tribrid– its numerous improvements enable ETD data acquisition with higher sensitivity, and at high resolution and high mass accuracy, which will make spectral assignments more reliable. The proper bioinformatic tools for glycopeptide analysis are still in development. An iterative analysis combining HCD and ETD data would be highly desirable.
References
Helenius, A., Aebi, M.: Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 (2004)
Varki, A.: Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3, 97–130 (1993)
Haltiwanger, R.S., Lowe, J.B.: Role of glycosylation in development. Annu. Rev. Biochem. 73, 491–537 (2004)
Rudd, P.M., Elliott, T., Cresswell, P., Wilson, I.A., Dwek, R.A.: Glycosylation and the immune system. Science 291, 2370–2376 (2001)
Brockhausen, I.: Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep. 7, 599–604 (2006)
Arnold, J.N., Saldova, R., Hamid, U.M., Rudd, P.M.: Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics 8, 3284–3293 (2008)
Grigorian, A., Mkhikian, H., Li, C.F., Newton, B.L., Zhou, R.W., Demetriou, M.: Pathogenesis of multiple sclerosis via environmental and genetic dysregulation of Nglycosylation. Semin. Immunopathol. 34, 415–424 (2012)
Christiansen, M.N., Chik, J., Lee, L., Anugraham, M., Abrahams, J.L., Packer, N.H.: Cell surface protein glycosylation in cancer. Proteomics 14, 525–546 (2013)
Scott, D.W., Patel, R.P.: Endothelial heterogeneity and adhesion molecules Nglycosylation: implications in leukocyte trafficking in inflammation. Glycobiology 23, 622–633 (2013)
Stuchlova Horynova, M., Raska, M., Clausen, H., Novak, J.: Aberrant O-glycosylation and anti-glycan antibodies in an autoimmune disease IgA nephropathy and breast adenocarcinoma. Cell. Mol. Life Sci. 70, 829–839 (2013)
Venkatakrishnan, V., Packer, N.H., Thaysen-Andersen, M.: Host mucin glycosylation plays a role in bacterial adhesion in lungs of individuals with cystic fibrosis. Expert Rev. Respir. Med. 7, 553–576 (2013)
Schedin-Weiss, S., Winblad, B., Tjernberg, L.O.: The role of protein glycosylation in Alzheimer disease. FEBS J. 281, 46–62 (2014)
Hägglund, P., Matthiesen, R., Elortza, F., Højrup, P., Roepstorff, P., Jensen, O.N., Bunkenborg, J.: An enzymatic deglycosylation scheme enabling identification of core fucosylated N-glycans and O-glycosylation site mapping of human plasma proteins. J. Proteome Res. 6, 3021–3031 (2007)
Zhang, H., Li, X.J., Martin, D.B., Aebersold, R.: Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 (2003)
Nilsson, J., Rüetschi, U., Halim, A., Hesse, C., Carlsohn, E., Brinkmalm, G., Larson, G.: Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 (2009)
Halim, A., Nilsson, J., Rüetschi, U., Hesse, C., Larson, G.: Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol. Cell. Proteomics (2012). doi:10.1074/mcp.M111.013649
Halim, A., Rüetschi, U., Larson, G., Nilsson, J.: LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J. Proteome Res. 12, 573–584 (2013)
Steentoft, C., Vakhrushev, S.Y., Vester-Christensen, M.B., Schjoldager, K.T., Kong, Y., Bennett, E.P., Mandel, U., Wandall, H., Levery, S.B., Clausen, H.: Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 (2011)
Vakhrushev, S.Y., Steentoft, C., Vester-Christensen, M.B., Bennett, E.P., Clausen, H., Levery, S.B.: Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol. Cell. Proteomics 12, 932–944 (2013)
Steentoft, C., Vakhrushev, S.Y., Joshi, H.J., Kong, Y., Vester-Christensen, M.B., Schjoldager, K.T., Lavrsen, K., Dabelsteen, S., Pedersen, N.B., Marcos-Silva, L., Gupta, R., Bennett, E.P., Mandel, U., Brunak, S., Wandall, H.H., Levery, S.B., Clausen, H.: Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488 (2013)
Yang, Z., Halim, A., Narimatsu, Y., Jitendra Joshi, H., Steentoft, C., Schjoldager, K.T., Alder Schulz, M., Sealover, N.R., Kayser, K.J., Paul Bennett, E., Levery, S.B., Vakhrushev, S.Y., Clausen, H.: The GalNAc-type O-Glycoproteome of CHO cells characterized by the SimpleCell strategy. Mol. Cell. Proteomics 13, 3224–3235 (2014)
Campos, D., Freitas, D., Gomes, J., Magalhães, A., Steentoft, C., Gomes, C., Vester-Christensen, M.B., Ferreira, J.A., Afonso, L.P., Santos, L.L., Pinto de Sousa, J., Mandel, U., Clausen, H., Vakhrushev, S.Y., Reis, C.A.: Probing the O-glycoproteome of gastric cancer cell lines for biomarker discovery. Mol. Cell. Proteomics 14, 1616–1629 (2015)
Trinidad, J.C., Schoepfer, R., Burlingame, A.L., Medzihradszky, K.F.: N- and O-glycosylation in the murine synaptosome. Mol. Cell. Proteomics (2013). doi:10.1074/mcp.M113.030007
Medzihradszky, K.F., Kaasik, K., Chalkley, R.J.: Tissue-specific glycosylation at the glycopeptide level. Mol. Cell. Proteomics 14, 2103–2110 (2015)
Darula, Z., Medzihradszky, K.F.: Affinity enrichment and characterization of mucin core-1 type glycopeptides from bovine serum. Mol. Cell. Proteomics 8, 2515–2526 (2009)
Darula, Z., Sherman, J., Medzihradszky, K.F.: How to dig deeper? Improved enrichment methods for mucin core-1 type glycopeptides. Mol. Cell. Proteomics (2012). doi:10.1074/mcp.O111.016774
Darula, Z., Chalkley, R.J., Lynn, A., Baker, P.R., Medzihradszky, K.F.: Improved identification of O-linked glycopeptides from ETD data with optimized scoring for different charge states and cleavage specificities. Amino Acids 41, 321–328 (2011)
Zubarev, R.A., Horn, D.M., Fridriksson, E.K., Kelleher, N.L., Kruger, N.A., Lewis, M.A., Carpenter, B.K., McLafferty, F.W.: Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 72, 563–573 (2000)
Syka, J.E.P., Coon, J.J., Schroeder, M.J., Shabanowitz, J., Hunt, D.F.: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 101, 9528–9533 (2004)
Good, D.M., Wirtala, M., McAlister, G.C., Coon, J.J.: Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6, 1942–1951 (2007)
Brockhausen, I., Schachter, H., Stanley, P.: O-GalNAc glycans. In: Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E. (eds.) Essentials of Glycobiology. 2nd edition. Chapter 9. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (2009)
Halim, A., Brinkmalm, G., Ruetschi, U., Westman-Brinkmalm, A., Portelius, E., Zetterberg, H., Blennow, K., Larson, G., Nilsson, J.: Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U. S. A. 108, 11848–11853 (2011)
Bern, M., Kil, Y.J., Becker, C.: Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics. Chapter 13:Unit13.20 (2012)
Domon, B., Costello, C.E.: A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J. 5, 397–409 (1988)
Baker, P.R., Trinidad, J.C., Chalkley, R.J.: Modification site localization scoring integrated into a search engine. Mol. Cell. Proteomics (2011). doi:10.1074/mcp.M111.008078
Yamada, K., Hyodo, S., Kinoshita, M., Hayakawa, T., Kakehi, K.: Hyphenated technique for releasing and MALDI MS analysis of O-glycans in mucin-type glycoprotein samples. Anal. Chem. 82, 7436–7443 (2010)
Yabu, M., Korekane, H., Miyamoto, Y.: Precise structural analysis of O-linked oligosaccharides in human serum. Glycobiology 24, 542–553 (2014)
Alpert, A.J.: Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 80, 62–76 (2008)
Wiśniewski, J.R., Zougman, A., Nagaraj, N., Mann, M.: Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009)
Trinidad, J.C., Barkan, D.T., Gulledge, B.F., Thalhammer, A., Sali, A., Schoepfer, R., Burlingame, A.L.: Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 11, 215–229 (2012)
Medzihradszky, K.F., Kaasik, K., Chalkley, R.J.: Characterizing sialic acid variants at the glycopeptide level. Anal. Chem. 87, 3064–3071 (2015)
Saba, J., Dutta, S., Hemenway, E., Viner, R.: Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int. J. Proteomics (2012). doi:10.1155/2012/560391
Baker, P.R., Chalkley, R.J.: MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 (2014)
Hortin, G.L., Sviridov, D., Anderson, N.L.: High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 54, 1608–1616 (2008)
Hao, P., Guo, T., Sze, S.K.: Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with electrostatic repulsion hydrophilic interaction chromatography. PLoS One (2011). doi:10.1371/journal.pone.0016884
de Jong, E.P., Griffin, T.J.: Online nanoscale ERLIC-MS outperforms RPLC-MS for shotgun proteomics in complex mixtures. J. Proteome Res. 11, 5059–5064 (2012)
Hao, P., Ren, Y., Dutta, B., Sze, S.K.: Comparative evaluation of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) and high-pH reversed phase (Hp-RP) chromatography in profiling of rat kidney proteome. J. Proteomics 82, 254–262 (2013)
Li, Q., Jain, M.R., Chen, W., Li, H.: A multidimensional approach to an in-depth proteomics analysis of transcriptional regulators in neuroblastoma cells. J. Neurosci. Methods 216, 118–127 (2013)
Loroch, S., Schommartz, T., Brune, W., Zahedi, R.P., Sickmann, A.: Multidimensional electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) for quantitative analysis of the proteome and phosphoproteome in clinical and biomedical research. Biochim. Biophys. Acta 1854, 460–468 (2015)
Sok Hwee Cheow, E., Hwan Sim, K., de Kleijn, D., Neng Lee, C., Sorokin, V., Sze, S.K.: Simultaneous enrichment of plasma soluble and extracellular vesicular glycoproteins using prolonged ultracentrifugation-ERLIC approach. Mol. Cell. Proteomics 14, 1657–1671 (2015)
Chang, C.F., Pan, J.F., Lin, C.N., Wu, I.L., Wong, C.H., Lin, C.H.: Rapid characterization of sugar-binding specificity by in-solution proximity binding with photosensitizers. Glycobiology 21, 895–902 (2011)
Tachibana, K., Nakamura, S., Wang, H., Iwasaki, H., Tachibana, K., Maebara, K., Cheng, L., Hirabayashi, J., Narimatsu, H.: Elucidation of binding specificity of Jacalin toward O-glycosylated peptides: quantitative analysis by frontal affinity chromatography. Glycobiology 16, 46–53 (2006)
Windwarder, M., Yelland, T., Djordjevic, S., Altmann, F.: Detailed characterization of the O-linked glycosylation of the neuropilin-1 c/MAM-domain. Glycoconj. J. (2015)
Acknowledgments
The authors are grateful to Andrew Alpert for the generous gift of the PolyWAX column, Samuel Myers for the WGA-POROS column, ProteinMetrics Inc. for access to the Byonic software. We would like to thank Agnes Arva for technical assistance. We appreciate the help of Robert Chalkley in editing our manuscript. This work was supported by the following grants: National Institutes of Health NIGMS 8P41GM103481, Hungarian Scientific Research Fund 105611 (to Z. Darula), National Research, Development and Innovation Fund BAROSS-DA07-DA-ESZK-07-2008-0036 (to the Biological Research Centre, HAS, director: P. Ormos), New Szechenyi Plan GOP-1.1.1-11-2012-0452, and by the Howard Hughes Medical Institute (to the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF, Director: A. L. Burlingame). Z. Darula was supported by the Janos Bolyai Fellowship of the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(PDF 584 kb)
Rights and permissions
About this article

Cite this article
Darula, Z., Sarnyai, F. & Medzihradszky, K.F. O-glycosylation sites identified from mucin core-1 type glycopeptides from human serum. Glycoconj J 33, 435–445 (2016). https://doi.org/10.1007/s10719-015-9630-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-015-9630-6