
Abstract
Autoimmune diseases such as multiple sclerosis (MS) result from complex and poorly understood interactions of genetic and environmental factors. A central role for T cells in MS is supported by mouse models, association of the major histocompatibility complex region, and association of critical T cell growth regulator genes such as interleukin-2 receptor (IL-2RA) and interleukin-7 receptor (IL-7RA). Multiple environmental factors (vitamin D3 deficiency and metabolism) converge with multiple genetic variants (IL-7RA, IL-2RA, MGAT1, and CTLA-4) to dysregulate Golgi N-glycosylation in MS, resulting in T cell hyperactivity, loss of self-tolerance and in mice, a spontaneous MS-like disease with neurodegeneration. Here, we review the genetic and biological interactions that regulate MS pathogenesis through dysregulation of N-glycosylation and how this may enable individualized therapeutic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is an autoimmune and neurodegenerative disorder of the central nervous system (CNS) characterized by inflammatory demyelination, axonal degeneration, and neuron loss [1–3]. Although mouse models of MS, such as experimental autoimmune encephalomyelitis (EAE), provide pathogenic insights, their relevance to MS is indirect. For example, MOG-induced EAE in C57BL/6 mice is monophasic and may more closely mimic the non-relapsing demyelinating disease acute disseminated encephalomyelitis rather than MS. Even “relapsing models” such as PLP-induced EAE in SJL mice do not closely recapitulate relapsing MS. Relapses in MS are separated by months and afflict new areas of the CNS, whereas SJL EAE relapses are recurring episodes of motor weakness separated by days. A direct approach to define pathogenic mechanisms in MS would delineate how known disease risk factors function and interact at the molecular level, utilizing mouse models such as EAE to confirm pathogenic mechanisms.
As with other complex trait diseases, multiple genetic and environmental factors combine to influence disease risk in MS and many other human autoimmune disorders, including systemic lupus erythematosus and type 1 diabetes (T1D) [4, 5]. Epidemiological studies indicate that MS risk is influenced by gender, sex hormones, ethnic origin, continental location/latitude/distance from the equator, smoking, viral exposure (e.g., Epstein–Barr virus), and vitamin D3 status [6–9]. As vitamin D3 is synthesized from 7-dehydrocholesterol in the skin following ultraviolet light exposure, a link between latitude, hours of sunshine, and vitamin D3 status has been made. This connection is supported by the observation that migration from high-risk areas with limited hours of sunlight to low-risk areas with greater hours of sunshine prior to puberty affords some protection against MS [10]. The latent viral or infectious hypothesis has been proposed, but all attempts at isolating and proving a causal role for a pathogen have failed, suggesting that pathogens act through molecular mimicry to drive pathogenic auto-reactive T cells.
A definitive role for genetics in MS was first demonstrated in elegant family studies by George Ebers and colleagues, where it was observed that first-degree relatives and identical twins display ~20−40- and ~300-fold increased risk over the general population, respectively [4]. Candidate gene studies have validated association of MS with genes in the major histocompatibility complex (MHC) region [11]. In African American patients, it was determined that the primary association was with the DRB1 gene, which was subsequently confirmed in large cohorts of patients of European descent [12, 13]. More recently, genome-wide association studies (GWAS) have identified approximately 50 potential genes associated with MS [14, 15]. A number of these genes also associate with other autoimmune diseases, such as IL7RA and IL2RA in T1D [16–18]. While the IL-7 and IL-2 pathways have previously been demonstrated to regulate autoimmunity and EAE in animal models [19, 20], such data are lacking for many of the other MS-associated variants. Validation as true MS risk factors requires much more than statistical association; rather, functional characterization of the changes induced by the polymorphism and evidence for pathogenicity of the same molecular pathway in animal models is necessary.
Although GWAS has identified approximately 50 genetic loci associated with MS, many critical issues remain. First, whether the detected polymorphism alters the biology of the nearest gene, as often assumed and labeled as such, or a more distant unrelated gene is left unresolved. Similarly, whether the detected variant is causal in disease or simply in linkage disequilibrium (LD) with a distant undetected causal polymorphism is also not addressed. The critical importance of these issues was best demonstrated by a GWAS of sickle cell anemia [21], a disease where the single genetic variant that induces disease has been established for many years. Despite this, the GWAS identified 179 non-causal polymorphisms with genome-wide significance that encompassed a 2.5-Mb region harboring multiple LD blocks and dozens of non-disease-related genes. Thus, many irrelevant variants and genes may be identified by GWAS analysis. A second critical issue of GWAS studies is the missing heritability. Except for the human leukocyte antigen (HLA), the identified variants in MS confer relatively small increments in disease risk and explain only ~20 % of the genetic variance that we know exists [15, 22], questioning the source of this missing heritability. Many explanations have been suggested including additional common variants with smaller effects or rare and highly penetrant variants that are overlooked in the current genome-wide arrays that are restricted to variants with allele frequencies of ≥5 % [21–24]. However, rare and highly penetrant variants often underlie disorders with Mendelian-type inheritance that have little or no environmental influences (e.g., cystic fibrosis). The fact that comprehensive loci analyses to date have not accounted for the predicted genetic variation in complex trait diseases suggests that the resolution of this dilemma lies in the complexity of the underlying genetics.
Susceptibility to complex trait diseases is multifactorial and results from the interactions of multiple contributing genes and environmental factors, each with potential to interact in nonlinear ways. Epistatic interactions, where two or more independent variants promote disease only when combined [25], are likely to go undetected by genetic screens such as GWAS that examine for point association. Evidence consistent with epistatic interactions in autoimmune disease has been reported in both humans and mice [26–28]. Moreover, MS concordance rates in monozygotic twins are only ~30 % [4], implying direct environmental impact on genetic risk. Indeed, Baranzini et al. have recently reported that there is no evidence for genetic, epigenetic, or transcriptome sequence differences that explain disease discordance in monozygotic twins discordant for MS [29]. It is interesting to note that all twin pairs studied had identical genotypes within the HLA loci and only one of the three twin pairs had DRB1*1501, a genetic variant with the strongest association with MS.
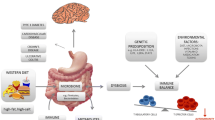
Despite the identification of multiple environmental and genetic risk factors for MS, there appears to be no obvious shared molecular mechanisms, although most appear immune related [15]. Single-gene disorders displaying Mendelian inheritance disrupt molecular pathways at a single step. However, a similar degree of pathway disruption may also be obtained through small defects in multiple genes within a single pathway. Thus, complex trait diseases like MS may arise from epistatic and/or additive interactions between multiple seemingly unrelated alleles and environmental factors that converge to dysregulate a critical final common pathway. Indeed, we recently reported that multiple environmental factors (vitamin D3 deficiency and metabolism) converge with multiple genetic variants (IL-7RA, IL-2RA, MGAT1, and CTLA-4) to dysregulate Golgi N-glycosylation in MS. Defective N-glycosylation of the T cell receptor (TCR) and cytotoxic T lymphocyte antigen 4 (CTLA-4) induces T cell hyperactivity, promotes loss of self-tolerance and in mice, induces a spontaneous MS-like disease [30–33]. Here, we review the genetic and biological interactions that differentially regulate MS risk through dysregulation of N-glycosylation, how this may promote pathogenesis, and the potential for individualized approaches to diagnostics and treatment (Fig. 1).

Multiple risk factors decrease N-glycan branching to promote diverse pathogenic mechanisms in multiple sclerosis. Human and mouse data indicate that genetic factors, the environment, and cytokines combine to decrease N-glycan branching. This in turn leads to multiple pathogenic mechanisms in multiple sclerosis (MS), including T cell hyperactivity, altered antigen-presenting cell (APC) function, and enhanced susceptibility to neurodegeneration. Recent mouse data also support a potential negative role for N-glycan branching in Treg suppressor function and re-myelination by oligodendrocyte precursor cells. Thus, defective N-glycan branching in MS results from multiple inputs, which in turn results in multiple phenotypic outputs that likely drive MS pathogenesis
N-glycosylation and regulatory mechanisms of growth and differentiation
The majority of cell surface receptors and transporters are modified by co-translational addition of asparagine (N)-linked glycans in the endoplasmic reticulum, with further modifications in the Golgi secretory pathway [34, 35]. Cell surfaces and the extracellular matrix with which they interact are heavily glycosylated, and the size, abundance, and complexity of these glycan structures provide information encoding distinct from the genome [36]. In contrast to proteins and nucleic acids, production of complex carbohydrates is not template driven, but rather depends on enzymatic activities and metabolic supply of substrates. Glycoprotein concentrations at the cell surface can be differentially regulated according to their affinities for the galectin family of endogenous lectins [30, 31, 37]. Galectins are ubiquitously expressed at the cell surface and extracellular matrix and interact with multivalent glycan ligands to form a molecular “lattice” at the cell surface [31, 38, 39]. The minimal binding structure for galectins is N-acetyllactosamine (Galactose β1,4N-acetylglucosamine) [40], with binding avidity to glycoproteins increasing in proportion to the number of N-glycans per protein (gene-encoded) and the degree of branching/structural modifications per N-glycan (context/environment dependent) [36]. N-glycan branching produced in the Golgi is dependent upon the sequential yet incomplete action of the Golgi α-mannosidases and N-acetylglucosaminyltransferases I, II, IV, and V (encoded by Mgat1, 2, 4, and 5), along with hexosamine pathway production of the substrate uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) [30, 41, 42]. Growth-promoting receptors frequently have high numbers of N-glycans (n > 5), while growth inhibitory receptors frequently have few N-glycans (n ≤ 4). This allows differential association with the galectin lattice dependent on Golgi branching activity, thereby regulating cellular transitions from growth to arrest [30]. This paradigm has been demonstrated for TCR/CTLA-4 in T cells and receptor tyrosine kinases/transforming growth factor-β receptor (TβR) in epithelial cells [30].
The intricate interplay between growth stimulatory and inhibitory signals shapes the T cell immune response and is critical for T cell tolerance. Golgi-mediated changes in N-glycan branching differentially control cell surface retention and endocytosis rates of glycoproteins, and in this manner, the galectin–glycoprotein lattice appears to incorporate both genetic and metabolic cues to control cellular function and cell fate decisions.
N-glycosylation and T cell-mediated autoimmunity in mice
In mice, targeted deficiencies of factors that inhibit growth of naïve T cells, such as CTLA-4, TβR, and regulatory T cells (Treg), result in spontaneous autoimmunity. Similarly, human autoimmunity is often associated with risk factors that control T cell growth, including the MHC region, CTLA-4 Thr17Ala (rs231775), IL2RA*T (rs2104286), IL7RA*C (rs6897932), and vitamin D3 deficiency. N-glycan branching is also a critical negative regulator of T cell growth and when genetically disrupted in mice, results in spontaneous autoimmunity [43]. Antigen independent and antigen-induced TCR clustering and signaling are both suppressed by galectin interactions with the TCR via N-glycans, thereby suppressing both basal and activation signaling [32]. IL-2 and IL-7, two well-described enhancers of T cell growth, regulate mRNA expression of multiple Golgi genes to suppress N-glycan branching and thereby enhance ligand-induced TCR clustering and signaling [44]. After cell division, N-glycan branching increases in T cell blasts, promoting cell surface retention of CTLA-4 to induce growth arrest [30].
After growth arrest, T cells differentiate into pro-inflammatory T helper 1 (Th1)/T helper 17 (Th17) cells, anti-inflammatory T helper 2 (Th2) cells, and/or induced T regulatory cells (iTreg). Th2 cells secrete IL-4, IL-5, IL-10, and IL-13 and provide host defense against extracellular pathogens, assist B cells and humoral immunity, and are generally anti-inflammatory. Th1 and Th17 cells are pro-inflammatory effector cells that secrete IFN-γ and IL-17, respectively, and have been shown to independently promote autoimmunity [45]. iTregs strongly inhibit growth of other T cells and are crucial in downregulation of T cell responses. The relative balance of these different cell types dictates inflammatory, allergic, and autoimmune responses. Deficiencies in N-glycan branching promote Th1 and Th17 responses over Th2 responses [46, 47] (Araujo and Demetriou, unpublished data).
In summary, N-glycan branching is a critical negative regulator of T cell growth, is directly downregulated by cytokines (IL-2 and IL-7) that enhance growth, and inhibits pro-inflammatory Th1/Th17 responses. Not surprisingly, genetic deficiencies in N-glycan branching in mice promote spontaneous autoimmunity. For example, mice deficient in Mgat5 develop spontaneous autoimmune kidney disease and display increased sensitivity to EAE [31]. Furthermore, significant differences in N-glycan branching and Golgi enzyme activity are observed among inbred mouse strains, with strains susceptible to EAE displaying defective N-glycan branching in T cells [33]. The PL/J strain, with the lowest levels of N-glycan branching, contains natural deficiencies in multiple N-glycan branching enzymes (i.e., Mgat1, 2, and 5) as demonstrated by mass spectroscopy and enzyme assays. PL/J mice with targeted deficiency in Mgat5 develop a spontaneous, late-onset clinical MS-like disease manifested by inflammatory demyelination and neurodegeneration [33]. A much milder form of disease is observed in wild-type PL/J mice, consistent with the defective N-glycan branching inherent to this inbred strain.
Autoimmunity and defective N-glycosylation in non-T cells
Data in mice suggest that defective N-glycosylation may also promote autoimmunity through dysfunction of non-T cells (Fig. 1). For example, deficiencies in the galectin–glycoprotein lattice also alter antigen-presenting cell (APC) function. Defective N-glycan branching and blockade of polylactosamine synthesis, which both weaken the galectin lattice, increase sensitivity to cytokine signaling and lower antigen-presenting cell activation thresholds [37, 48], consistent with a regulatory role for N-glycosylation in tolerogenic signaling in APCs. Indeed, galectin-1, through binding to cell surface glycans and strengthening the galectin lattice, induce tolerogenic dendritic cells that secrete IL-27 to promote IL-10-mediated T cell tolerance and suppress EAE [49].
Defective N-glycosylation may also promote autoimmunity through molecular mechanisms distinct from the galectin–glycoprotein lattice. Spontaneous autoimmunity in mice deficient in Golgi alpha-mannosidase-II (αM-II) is associated with minimal reductions in N-glycan branching in T cells but marked deficiencies in other tissues such as the kidney and red blood cells [50]. αM-II deficiency induces a systemic lupus erythematosus-like syndrome in mice characterized by elevated systemic anti-nuclear antibody titers, dyserythropoietic anemia, glomerular deposition of immunoglobulins and complement component C3, and glomerulonephritis leading to sclerosis, renal dysfunction, and kidney failure [51]. Jamey Marth and colleagues have proposed that αM-II deficiency induced increases in cell surface mannose exposed N-glycans hyperactivates an innate immune response through binding to mannose-binding lectin receptors [51]. Mannose exposed N-glycans are normally only seen at high density in pathogens [52], with increased levels from αM-II deficiency potentially resulting in a defect in self-tolerance by innate immune cells and chronic activation.
Organ-specific autoimmune diseases such as MS may also be influenced by increased sensitivity of target cells to death. For example, in addition to inflammatory demyelination, MS is characterized by neuron loss and axonal damage even in the absence of inflammation. Consistent with this, Mgat5 deficiency in PL/J mice results not only in spontaneous inflammatory demyelination but also neurodegeneration, characterized by neuronal loss and axonal damage in both inflamed and non-inflamed CNS tissue [33]. Moreover, targeted deficiency of Mgat1 in neurons induces their apoptosis in vivo, confirming that N-glycan branching directly regulates neuronal viability [53]. These data suggest that N-glycan branching independently promotes both T cell-mediated autoimmunity and neurodegeneration, two hallmarks of MS.
Environmental regulation of autoimmunity via N-glycosylation
N-glycan branching in T cells is directly influenced by metabolism and vitamin D3, thereby providing a molecular mechanism for environmental regulation of T cell-mediated autoimmunity. The N-glycan branching enzymes (Mgat1, 2, 4, and 5) all utilize the same sugar-nucleotide donor, namely UDP-GlcNAc, but do so with declining efficiency [36]. The Km of Mgat4 and Mgat5 for UDP-GlcNAc is ~5 and ~11 mM, respectively, whereas the Golgi concentration of UDP-GlcNAc is only ~1.5 mM. Thus, these enzymes are under-saturated for UDP-GlcNAc, and small changes in UDP-GlcNAc concentration can lead to significant changes in N-glycan branching, T cell growth/differentiation, and autoimmunity [30, 41].
De novo synthesis of UDP-GlcNAc by the hexosamine pathway requires highly regulated intermediates of carbohydrate, nitrogen, and fatty acid metabolism [41], and in this manner, N-glycan branching is sensitive to metabolic status and the nutrient environment of the cell. Indeed, increased supply of glucose, glutamine (a critical nitrogen metabolite), or acetyl-CoA (the final metabolite of free fatty acids) enhances N-glycan branching in T cells in vitro. UDP-GlcNAc may also be synthesized through salvage of the monosaccharides glucosamine (GlcN) and N-acetylglucosamine (GlcNAc). However, unlike GlcNAc, GlcN may also be shunted into glycolysis and ATP production. Indeed, when titrated in culture, GlcN first increases then decreases N-glycan branching in T cells [41]. In contrast, GlcNAc cannot enter glycolysis, is not metabolized, and is observed to only enhance N-glycan branching [41, 54]. Indeed, GlcNAc supplementation in vitro and/or in vivo suppresses T cell growth by limiting TCR signaling and enhancing CTLA-4 surface retention, inhibits Th1 and Th17 responses, and suppresses EAE as well as autoimmune diabetes [41, 47]. Moreover, Murch et al. observed that oral GlcNAc therapy inhibited clinical disease in 8 of 12 children with treatment-resistant inflammatory bowel disease [55]. Thus, metabolism regulates N-glycan branching and thereby influences susceptibility to T cell-mediated autoimmunity in mice.
Vitamin D3 deficiency is a well-described environmental risk factor associated with MS that we have recently shown to regulate N-glycan branching in T cells to suppress growth and EAE. Previous epidemiological investigations revealed that MS risk increases with distance from the equator and the corresponding decline in ultraviolet exposure [56, 57]. Vitamin D3 is synthesized from 7-dehydrocholesterol in the skin upon ultraviolet sun exposure, and its deficiency strongly associates with MS [8, 58, 59]. 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3), the active form of vitamin D3, inhibits T cell activation, Th1 differentiation, and suppresses EAE in mice by acting on T cells [60–62], yet molecular mechanisms have been unclear.
1,25(OH)2D3 increases N-glycan branching in activated ex vivo T cells to suppress their growth [44]. Reducing dietary supply of vitamin D3 in mice decreased N-glycan branching in T cells, whereas intraperitoneal injection of 1,25(OH)2D3 increased N-glycan branching. Myelin basic protein-induced EAE was inhibited by intraperitoneal injection of 1,25(OH)2D3 in the absence but not presence of swainsonine, an inhibitor of N-glycan branching. Combined, these data suggest that vitamin D3 suppresses T cell growth and EAE by enhancing N-glycan branching in T cells.
In summary, two independent environmental factors, namely metabolism/nutrient supply and sunshine/vitamin D3, influence T cell-mediated autoimmunity by regulating N-glycan branching (Fig. 1). Metabolic homeostasis consists of multiple feedback mechanisms, yet small changes in homeostatic set points with age and environmental cues can be clinically important in complex trait diseases such as MS. Therapeutic intervention with oral GlcNAc and/or vitamin D3 may provide a simple treatment to enhance N-glycan branching and suppress MS.
Genetic and environmental dysregulation of N-glycosylation in multiple sclerosis
Multiple genetic and environmental risk factors have been linked to MS; however, defining how these combine at the molecular level to promote disease has been a great challenge. The data described above define a critical role for environmental and genetic dysregulation of N-glycan branching in mouse T cells and autoimmunity, suggesting similar mechanisms may be relevant to human T cells and MS. Indeed, our group recently reported that multiple environmental factors (sunlight/vitamin D3 and metabolism) converge with multiple genetic variants (IL-7RA, IL-2RA, MGAT1, and CTLA-4) to dysregulate N-glycosylation in MS [44].
The IL2RA*T (rs2104286) and IL7RA*C (rs6897932) MS risk alleles are the common alleles in Caucasian populations (frequency ~75 %) and are associated with enhanced secretion of soluble receptors that block signaling by cognate cytokines [7, 16, 44, 63–66]. We observed that IL-2 and IL-7 are critical regulators of N-glycan branching, thereby controlling T cell growth [44, 67]. Consistent with this, soluble receptors associated with the IL2RA*T and IL7RA*C MS risk variants downregulate MGAT1 mRNA and N-glycan branching in human T cell blasts (Fig. 1). As these two MS risk variants directly regulated MGAT1, targeted sequencing of the human MGAT1 gene was undertaken. An MS-associated haplotype of MGAT1 (IVA and VT-T polymorphisms; rs7726005, rs2070924, and rs2070925) was identified that reduced or enhanced N-glycan branching depending on metabolism and UDP-GlcNAc supply to the Golgi. The MGAT1 IVAVT-T haplotype enhances mRNA levels and enzyme activity ~2−3-fold, thereby increasing the N-glycan product of Mgat1 while also limiting UDP-GlcNAc supply to downstream Mgat4 and 5. Mgat1, 2, 4, and 5 act in a sequential manner but with declining efficiency as enzyme levels and catalytic efficiencies of UDP-GlcNAc utilization decrease in the same order. The Km of Mgat4 and 5 for UDP-GlcNAc is significantly worse than Mgat1 (~5 and ~11 mM versus ~0.04 mM, respectively); allowing increased Mgat1 protein to outcompete Mgat4 and 5 for UDP-GlcNAc in the medial Golgi [30]. Thus, under basal UDP-GlcNAc levels (~1.5 mM), the MGAT1 IVAVT-T haplotype functions dominantly to reduce N-glycan branching. However, with increasing UDP-GlcNAc and/or Mgat5 levels, enhanced Mgat1 expression is not as effective in limiting supply of UDP-GlcNAc to Mgat4 and 5, allowing Mgat4 and 5 to act upon the increased supply of N-glycan acceptors from MGAT1 IVAVT-T, resulting in enhanced N-glycan branching. Thus, the phenotypic effect of the MS-associated MGAT1 IVAVT-T haplotype directly depends on metabolic status of the cell and production of UDP-GlcNAc; albeit basal UDP-GlcNAc conditions and reduced N-glycan branching are expected to predominate. Monozygotic twins are discordant for MS ~70 % of the time. The MGAT1 IVAVT-T haplotype provides an example of how the same genetic risk factor may both promote and inhibit MS conditional on the environment.
The MGAT1 IVAVT-T haplotype and the IL2RA*T and IL7RA*C MS risk alleles influence N-glycan branching by having opposing effects on Mgat1 expression. Consistent with this, upregulation of Mgat1 by IL-2 and/or IL-7 signaling enhances N-glycan branching when Mgat1 is suppressed by IL2RA*T and IL7RA*C but further decreases N-glycan branching when Mgat1 is already increased by the MGAT1 IVAVT-T haplotype [44]. In other words, upregulation of Mgat1 by IL-2 and/or IL-7 enhances or reduces branching depending on baseline Mgat1 activity, which differs based on the presence of the different MGAT1 variants. This provides a second conditional mechanism that controls N-glycan branching in MS.
Genetically induced downregulation of N-glycan branching in human T cell blasts is expected to reduce CTLA-4 surface retention and thereby promotes T cell growth [30]. The Thr17Ala polymorphism in the human CTLA-4 gene (49A/G, rs231775) encodes a signal peptide variant with inefficient glycosylation [68, 69]. This non-synonymous polymorphism associates with type 1 diabetes but not MS [70, 71], reduces average N-glycan occupancy at the two N-X-S/T sites from two to one, and decreases the number of branched N-glycans and CTLA-4 surface levels to enhance T cell growth (Fig. 1). The MGAT1 IVAVT-T haplotype also limits CTLA-4 surface levels when expressed with the common CTLA-4 allele (CTLA-4 Thr17; two N-glycans), whereas combining the MGAT1 IVAVT-T haplotype with the CTLA-4 Ala17 variant (one N-glycan) further reduces CTLA-4 surface levels [44]. CTLA-4 surface expression is restored by increasing UDP-GlcNAc levels with GlcNAc supplementation in all genotype combinations, confirming an additional mechanism regulated by metabolism.
In summary, the MGAT1 IVAVT-T haplotype lowers N-glycan branching, T cell activation thresholds, and CTLA-4 cell surface expression in a manner that is sensitive to metabolic conditions (i.e., UDP-GlcNAc), activity of other Golgi enzymes (e.g., Mgat5), the number of N-glycans attached to CTLA-4, and IL2/IL-7 signaling (Fig. 1).
These biological interactions predict specific genetic interactions in MS. Indeed, epistatic and additive interactions were observed between the four variants as expected [44]. The MGAT1 IVAVT-T haplotype increases MS risk when there are less than four copies of the IL2RA*T and IL7RA*C risk alleles, whereas no association is observed in the presence of four copies of the IL2RA*T and IL7RA*C variants, the latter consistent with opposing effects on Mgat1 expression optimizing Mgat1 activity and enhancing N-glycan branching. The MGAT1 IVAVT-T haplotype also significantly associated with MS in CTLA-4 Ala17 carriers (one N-glycan), but not CTLA-4 Thr17 homozygotes (two N-glycans). Moreover, the MGAT1 IVAVT-T haplotype promotes MS when there are less than six alleles of CTLA-4 Thr17, IL2RA*T, and IL7RA*C, whereas a marginally significant protective effect was observed with six alleles of CTLA-4 Thr17, IL2RA*T, and IL7RA*C [44]. The latter combination is expected to be protective as Mgat1 activity, N-glycan branching, and N-glycan number on CTLA-4 are optimized. Importantly, these genetic interactions are observed despite lack of point association and marginal effects of CTLA-4 Thr17Ala, indicative of epistatic interactions.
Vitamin D3 enhances N-glycan branching to suppress T cell growth and EAE in mice, while deficiency of vitamin D3 is associated with MS. To investigate possible interactions with genetic variants, we examined the effects of 1,25(OH)2D3 on human T cell blasts [44]. Remarkably, 1,25(OH)2D3 enhanced MGAT1 mRNA levels, similar to the MGAT1 IVAVT-T haplotype but opposite of the IL2RA*T and IL7RA*C risk alleles. Consistent with this effect on Mgat1, 1,25(OH)2D3 enhanced N-glycan branching in T cells with two or more copies of the IL2RA*T + IL7RA*C risk alleles (where Mgat1 expression is reduced). In contrast, N-glycan branching in T cells homozygous for the IL2RA*C and IL7RA*T protective alleles, where Mgat1 expression is not suppressed, was unchanged or reduced [44]. As a very small minority of Caucasians is homozygous for both the IL2RA*C + IL7RA*T protective alleles (~0.5 %), vitamin D3 deficiency is expected to reduce N-glycan branching in the majority of the Caucasian population.
IL2RA*T, IL7RA*C, CTLA-4 Ala17, and vitamin D3 deficiency also associate with T1D [16–18, 72]. The non-obese diabetic mouse is deficient in N-glycan branching in T cells, while oral GlcNAc is able to suppress development of autoimmune diabetes in these mice [33, 41]. Another independent variant of IL2RA (rs11594656) also associates with both MS and T1D, but paradoxically in opposite directions [16]. These data suggest that defective N-glycosylation also contributes to T1D risk.
Conclusions
Complex trait diseases such as MS develop from multifaceted and poorly understood interactions between genetics and the environment. While genetic and epidemiological studies have identified a number of genetic and environmental risk factors in MS, most appear to only marginally increase risk, do not account for all heritability, and display no obvious common molecular mechanism. Epistatic interactions, where two or more factors promote disease only when combined, are likely to go undetected in approaches assessing only point association such as GWAS. Here, we reviewed evidence suggesting that in MS, epistatic interactions between multiple independent genetic variants and environmental factors combine in a nonlinear fashion to dysregulate a common biochemical pathway, namely Golgi N-glycosylation. Each factor may only have a minor genetic or biological effect on risk and N-glycosylation, but specific combinations lead to more dramatic changes in N-glycan branching. Moreover, the same variant may either increase or decrease risk depending on co-inheritance of other variants and/or environmental factors. This paradigm suggests that future studies only examining point association, such as GWAS, are unlikely to adequately define heritability. Rather, molecular mechanistic studies of human variants enlightened by mouse data are likely required to intelligently and selectively examine for epistatic interactions and define disease mechanisms. For example, there are at least ~30 genes that alter N-glycan branching and may be screened for functional variants and epistatic interactions.
Defective N-glycosylation in MS results from multiple inputs, both environmental and genetic, but importantly also results in multiple phenotypic outputs (Fig. 1). Human and mouse data suggest that defective N-glycosylation contributes to MS by affecting multiple cell types and molecular mechanisms. In addition to defects in T cell growth and self-tolerance, defective N-glycosylation may also promote disease via hyperactive innate immune responses and increased sensitivity of neurons to death [37, 48, 49, 51, 53]. While the effects on T cells are defined in both mouse and humans, additional work is required to determine whether the genetic (e.g., MGAT1 IVAVT-T, IL2RA*T, and IL7RA*C) and/or environmental (e.g., vitamin D3 and UDP-GlcNAc metabolism) factors also directly alter innate immune activity and neurodegeneration in human cells via defective N-glycosylation. For example, the MGAT1 IVAVT-T haplotype increases the amount of mannose exposed N-glycans in peripheral blood monocytes. If this phenotype was also prominent in oligodendrocytes, exposure of these cryptic mannose residues may hyperactivate innate immune responses to promote demyelination.
Current treatment strategies for MS are predominated by injectable therapies with modest efficacy, high cost, and significant side effects, which can affect tolerability and compliance. The limitations of current medications warrant investigations into alternative therapeutic strategies, particularly those that directly target an underlying molecular mechanism promoting disease, rather than nonspecific immunomodulation and/or immunosuppression. Therapeutic supplementation of the Golgi to increase N-glycan biosynthesis may provide such a therapy. Both vitamin D3 and GlcNAc are orally active, reverse deficiencies in N-glycan branching in mice and humans, and inhibit EAE and spontaneous autoimmune diabetes in mice [41, 61, 73]. More recent data from our lab have shown that oral GlcNAc also inhibits Th1 and Th17 responses and disease progression in EAE when administered after disease onset [47]. A pilot study of oral GlcNAc in pediatric treatment-resistant inflammatory bowel disease reported that 8 out of 12 children with severe disease went into clinical remission with evidence of histological improvement [55]. Three of the responders relapsed within ~1 month following disruption of GlcNAc therapy, but improved again once therapy was reinitiated [55]. These data suggest that a human clinical trial of GlcNAc in MS is warranted.
References
Steinman L (2001) Multiple sclerosis: a two-stage disease. Nat Immunol 2:762–764
Filippi M, Rocca MA (2005) MRI evidence for multiple sclerosis as a diffuse disease of the central nervous system. J Neurol 252(Suppl 5):v16–v24
Pirko I, Lucchinetti CF, Sriram S, Bakshi R (2007) Gray matter involvement in multiple sclerosis. Neurology 68:634–642
Ebers GC, Bulman DE, Sadovnick AD, Paty DW, Warren S, Hader W, Murray TJ, Seland TP, Duquette P, Grey T et al (1986) A population-based study of multiple sclerosis in twins. N Engl J Med 315:1638–1642
Ebers GC, Sadovnick AD, Risch NJ (1995) A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 377:150–151
Oldstone MB (1987) Molecular mimicry and autoimmune disease. Cell 50:819–820
Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL (2007) Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357:851–862
Smolders J, Damoiseaux J, Menheere P, Hupperts R (2008) Vitamin D as an immune modulator in multiple sclerosis, a review. J Neuroimmunol 194:7–17
Compston A, Coles A (2002) Multiple sclerosis. Lancet 359:1221–1231
Kurtzke JF (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev 6:382–427
Lincoln MR, Montpetit A, Cader MZ, Saarela J, Dyment DA, Tiislar M, Ferretti V, Tienari PJ, Sadovnick AD, Peltonen L, Ebers GC, Hudson TJ (2005) A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet 37:1108–1112
Oksenberg JR, Barcellos LF, Cree BA, Baranzini SE, Bugawan TL, Khan O, Lincoln RR, Swerdlin A, Mignot E, Lin L, Goodin D, Erlich HA, Schmidt S, Thomson G, Reich DE, Pericak-Vance MA, Haines JL, Hauser SL (2004) Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet 74:160–167
Yeo TW, De Jager PL, Gregory SG, Barcellos LF, Walton A, Goris A, Fenoglio C, Ban M, Taylor CJ, Goodman RS, Walsh E, Wolfish CS, Horton R, Traherne J, Beck S, Trowsdale J, Caillier SJ, Ivinson AJ, Green T, Pobywajlo S, Lander ES, Pericak-Vance MA, Haines JL, Daly MJ, Oksenberg JR, Hauser SL, Compston A, Hafler DA, Rioux JD, Sawcer S (2007) A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann Neurol 61:228–236
Oksenberg JR, Baranzini SE (2010) Multiple sclerosis genetics—is the glass half full, or half empty? Nat Rev Neurol 6:429–437
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA et al (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476:214–219
Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, Clark PM, Healy B, Walker N, Aubin C, Oksenberg JR, Hauser SL, Compston A, Sawcer S, De Jager PL, Wicker LS, Todd JA, Hafler DA (2009) IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet 5:e1000322
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AA, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JM, Guja C, Ionescu-Tirgoviste C, Simmonds MJ, Heward JM, Gough SC, Dunger DB, Wicker LS, Clayton DG (2007) Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39:857–864
Malek TR (2008) The biology of interleukin-2. Annu Rev Immunol 26:453–479
Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL (1994) Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med 180:1955–1960
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB (2010) Rare variants create synthetic genome-wide associations. PLoS Biol 8:e1000294
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI (2008) Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am J Hum Genet 82:100–112
McClellan J, King MC (2010) Genetic heterogeneity in human disease. Cell 141:210–217
Culverhouse R, Suarez BK, Lin J, Reich T (2002) A perspective on epistasis: limits of models displaying no main effect. Am J Hum Genet 70:461–471
Gray-McGuire C, Moser KL, Gaffney PM, Kelly J, Yu H, Olson JM, Jedrey CM, Jacobs KB, Kimberly RP, Neas BR, Rich SS, Behrens TW, Harley JB (2000) Genome scan of human systemic lupus erythematosus by regression modeling: evidence of linkage and epistasis at 4p16–15.2. Am J Hum Genet 67:1460–1469
Sundvall M, Jirholt J, Yang HT, Jansson L, Engstrom A, Pettersson U, Holmdahl R (1995) Identification of murine loci associated with susceptibility to chronic experimental autoimmune encephalomyelitis. Nat Genet 10:313–317
Prins JB, Todd JA, Rodrigues NR, Ghosh S, Hogarth PM, Wicker LS, Gaffney E, Podolin PL, Fischer PA, Sirotina A et al (1993) Linkage on chromosome 3 of autoimmune diabetes and defective Fc receptor for IgG in NOD mice. Science 260:695–698
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell CJ, Kim RW, May GD, Woodward JE, Caillier SJ, McElroy JP, Gomez R, Pando MJ, Clendenen LE, Ganusova EE, Schilkey FD, Ramaraj T, Khan OA, Huntley JJ, Luo S, Kwok PY, Wu TD, Schroth GP, Oksenberg JR, Hauser SL, Kingsmore SF (2010) Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464:1351–1356
Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129:123–134
Demetriou M, Granovsky M, Quaggin S, Dennis JW (2001) Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409:733–739
Chen IJ, Chen HL, Demetriou M (2007) Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling. J Biol Chem 282:35361–35372
Lee SU, Grigorian A, Pawling J, Chen IJ, Gao G, Mozaffar T, McKerlie C, Demetriou M (2007) N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J Biol Chem 282:33725–33734
Schachter H (1991) The ‘yellow brick road’ to branched complex N-glycans. Glycobiology 1:453–461
Kornfeld R, Kornfeld S (1985) Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 54:631–664
Dennis JW, Nabi IR, Demetriou M (2009) Metabolism, cell surface organization, and disease. Cell 139:1229–1241
Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306:120–124
Brewer CF, Miceli MC, Baum LG (2002) Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol 12:616–623
Ahmad N, Gabius HJ, Andre S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF (2004) Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem 279:10841–10847
Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, Urashima T, Oka T, Futai M, Muller WE, Yagi F, Kasai K (2002) Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta 1572:232–254
Grigorian A, Lee SU, Tian W, Chen IJ, Gao G, Mendelsohn R, Dennis JW, Demetriou M (2007) Control of T cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J Biol Chem 282:20027–20035
Sasai K, Ikeda Y, Fujii T, Tsuda T, Taniguchi N (2002) UDP-GlcNAc concentration is an important factor in the biosynthesis of beta1,6-branched oligosaccharides: regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 12:119–127
Grigorian A, Torossian S, Demetriou M (2009) T-cell growth, cell surface organization, and the galectin–glycoprotein lattice. Immunol Rev 230:232–246
Mkhikian H, Grigorian A, Li CF, Chen HL, Newton B, Zhou RW, Beeton C, Torossian S, Tatarian GG, Lee SU, Lau K, Walker E, Siminovitch KA, Chandy KG, Yu Z, Dennis JW, Demetriou M (2011) Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun 2:334
Steinman L (2008) A rush to judgment on Th17. J Exp Med 205:1517–1522
Morgan R, Gao G, Pawling J, Dennis JW, Demetriou M, Li B (2004) N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol 173:7200–7208
Grigorian A, Araujo L, Naidu NN, Place DJ, Choudhury B, Demetriou M (2011) N-Acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J Biol Chem 286:40133–40141
Togayachi A, Kozono Y, Ishida H, Abe S, Suzuki N, Tsunoda Y, Hagiwara K, Kuno A, Ohkura T, Sato N, Sato T, Hirabayashi J, Ikehara Y, Tachibana K, Narimatsu H (2007) Polylactosamine on glycoproteins influences basal levels of lymphocyte and macrophage activation. Proc Natl Acad Sci U S A 104:15829–15834
Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, Geffner JR, Rabinovich GA (2009) Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol 10:981–991
Chui D, Sellakumar G, Green R, Sutton-Smith M, McQuistan T, Marek K, Morris H, Dell A, Marth J (2001) Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc Natl Acad Sci U S A 98:1142–1147
Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth JD (2007) Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity 27:308–320
Dam TK, Brewer CF (2010) Lectins as pattern recognition molecules: the effects of epitope density in innate immunity. Glycobiology 20:270–279
Ye Z, Marth JD (2004) N-glycan branching requirement in neuronal and postnatal viability. Glycobiology 14:547–558
Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24:2784–2799
Salvatore S, Heuschkel R, Tomlin S, Davies SE, Edwards S, Walker-Smith JA, French I, Murch SH (2000) A pilot study of N-acetyl glucosamine, a nutritional substrate for glycosaminoglycan synthesis, in paediatric chronic inflammatory bowel disease. Aliment Pharmacol Ther 14:1567–1579
Ramagopalan SV, Dyment DA, Ebers GC (2008) Genetic epidemiology: the use of old and new tools for multiple sclerosis. Trends Neurosci 31:645–652
Ascherio A, Munger KL, Simon KC (2010) Vitamin D and multiple sclerosis. Lancet Neurol 9:599–612
Noseworthy JH (1999) Progress in determining the causes and treatment of multiple sclerosis. Nature 399:A40–A47
Munger KL, Zhang SM, O'Reilly E, Hernan MA, Olek MJ, Willett WC, Ascherio A (2004) Vitamin D intake and incidence of multiple sclerosis. Neurology 62:60–65
Tsoukas CD, Provvedini DM, Manolagas SC (1984) 1,25-dihydroxyvitamin D3: a novel immunoregulatory hormone. Science 224:1438–1440
Lemire JM, Archer DC (1991) 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Investig 87:1103–1107
Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE (2011) 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol 41:822–832
Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, Caillier SJ, Ban M, Goris A, Barcellos LF, Lincoln R, McCauley JL, Sawcer SJ, Compston DA, Dubois B, Hauser SL, Garcia-Blanco MA, Pericak-Vance MA, Haines JL (2007) Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet 39:1083–1091
Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallstrom E, Khademi M, Oturai A, Ryder LP, Saarela J, Harbo HF, Celius EG, Salter H, Olsson T, Hillert J (2007) Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet 39:1108–1113
Rose T, Lambotte O, Pallier C, Delfraissy JF, Colle JH (2009) Identification and biochemical characterization of human plasma soluble IL-7R: lower concentrations in HIV-1-infected patients. J Immunol 182:7389–7397
Maier LM, Anderson DE, Severson CA, Baecher-Allan C, Healy B, Liu DV, Wittrup KD, De Jager PL, Hafler DA (2009) Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol 182:1541–1547
Grigorian A, Mkhikian H, Demetriou M (2012) Interleukin-2, interleukin-7, T cell-mediated autoimmunity, and N-glycosylation. Ann N Y Acad Sci. doi:10.1111/j.1749-6632.2011.06391.x
Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C (2002) A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem 277:46478–46486
Maurer M, Loserth S, Kolb-Maurer A, Ponath A, Wiese S, Kruse N, Rieckmann P (2002) A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics 54:1–8
Kavvoura FK, Ioannidis JP (2005) CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol 162:3–16
Bagos PG, Karnaouri AC, Nikolopoulos GK, Hamodrakas SJ (2007) No evidence for association of CTLA-4 gene polymorphisms with the risk of developing multiple sclerosis: a meta-analysis. Mult Scler 13:156–168
Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM (2001) Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 358:1500–1503
Zella JB, McCary LC, DeLuca HF (2003) Oral administration of 1,25-dihydroxyvitamin D3 completely protects NOD mice from insulin-dependent diabetes mellitus. Arch Biochem Biophys 417:77–80
Acknowledgments
Research was supported by the National Institutes of Health R01AI053331 and R01AI082266 to M.D. and F32AI081456 to A.G. through the National Institute of Allergy and Infectious Disease, F30 HL108451 to H.M. through the National Heart Lung and Blood Institute, as well as through a Collaborative Multiple Sclerosis Research Center Award to M.D.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Glycosylation and Immunity [34:3].
Rights and permissions
About this article
Cite this article
Grigorian, A., Mkhikian, H., Li, C.F. et al. Pathogenesis of multiple sclerosis via environmental and genetic dysregulation of N-glycosylation. Semin Immunopathol 34, 415–424 (2012). https://doi.org/10.1007/s00281-012-0307-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-012-0307-y