
Abstract
Fucosylation is an important type of glycosylation involved in cancer, and fucosylated proteins could be employed as cancer biomarkers. Previously, we reported that fucosylated N-glycans on haptoglobin in the sera of patients with pancreatic cancer were increased by lectin-ELISA and mass spectrometry analyses. However, an increase in fucosylated haptoglobin has been reported in various types of cancer. To ascertain if characteristic fucosylation is observed in each cancer type, we undertook site-specific analyses of N-glycans on haptoglobin in the sera of patients with five types of operable gastroenterological cancer (esophageal, gastric, colon, gallbladder, pancreatic), a non-gastroenterological cancer (prostate cancer) and normal controls using ODS column LC-ESI MS. Haptoglobin has four potential glycosylation sites (Asn184, Asn207, Asn211, Asn241). In all cancer samples, monofucosylated N-glycans were significantly increased at all glycosylation sites. Moreover, difucosylated N-glycans were detected at Asn 184, Asn207 and Asn241 only in cancer samples. Remarkable differences in N-glycan structure among cancer types were not observed. We next analyzed N-glycan alditols released from haptoglobin using graphitized carbon column LC-ESI MS to identify the linkage of fucosylation. Lewis-type and core-type fucosylated N-glycans were increased in gastroenterological cancer samples, but only core-type fucosylated N-glycan was relatively increased in prostate cancer samples. In metastatic prostate cancer, Lewis-type fucosylated N-glycan was also increased. These data suggest that the original tissue/cell producing fucosylated haptoglobin is different in each cancer type and linkage of fucosylation might be a clue of primary lesion, thereby enabling a differential diagnosis between gastroenterological cancers and non-gastroenterological cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycosylation is a critical post-translational modification of proteins. Fucosylation is an important event in glycosylation because it results in the formation of blood-type antigens and cancer-associated carbohydrate antigens [1]. Several researchers have reported that changes in glycan structures occur in pathologic conditions [2] and that the fucosylation of glycoproteins is associated with cancer and inflammation [3]. Hence, fucosylated target proteins have been identified [4] and considered to be potential tumor markers.
Previously, we reported that fucosylated N-glycans on haptoglobin in the sera of patients with pancreatic cancer were increased according to analyses by lectin-enzyme-linked immunosorbent assay (lectin-ELISA) and mass spectrometry [5, 6]. Haptoglobin is an acute-phase protein produced in the liver and contains four glycosylation sites [7, 8]. In healthy individuals, most haptoglobin is not fucosylated [9, 10] because a normal liver expresses low levels of fucosyltransferases and guanosine diphosphate fucose (GDP-Fuc, a common donor substrate for fucosyltransferases) [11]. Therefore, our report suggested that fucosylated N-glycans on haptoglobin could be novel tumor markers. Until now, N-glycans on haptoglobin have been analyzed by various methods in patients with various types of cancer: hepatocellular carcinoma [12–15], lung cancer [16–19], pancreatic cancer [20–23], colon cancer [24, 25], gastric cancer [26], ovarian cancer [27–29], prostate cancer [30–32] and breast cancer [33, 34]. However, though samples derived from the same type of cancer were analyzed, each result was different because of disparities in analytical strategy, including sample preparation and analytical method employed (e.g., lectin blotting, western blotting, liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS), high-performance liquid chromatography (HPLC) and capillary electrophoresis). To gain more accurate and comparable information of the N-glycan structures on haptoglobin (especially the fucosylation site and linkage) various types of cancer samples should be analyzed using the same analytical strategy.
To ascertain if characteristic fucosylation on haptoglobin is observed among different types of cancer, we undertook site-specific analyses of N-glycans on haptoglobin in the sera of patients with five types of operable gastroenterological cancers (esophageal, gastric, colon, gallbladder, pancreatic), a non-gastroenterological cancer (prostate) and normal controls. Furthermore, alditol N-glycans released from haptoglobin were analyzed using LC-ESI MS to identify and compare the linkage of fucosylation in each cancer sample. Identification of a characteristic fucosylation site and linkage in haptoglobin could be a novel type of cancer biomarker for the differential diagnosis of various types of cancer.
Materials and methods
The study protocol was approved by the ethics committees of participating hospitals, Osaka University (Osaka, Japan) and Hiroshima University (Hiroshima, Japan). All patients provided have written informed consent to be included in this study.
Lysylendopeptidase was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Sequencing-grade modified trypsin was obtained from Promega (Madison, WI, USA). Polyclonal rabbit anti-human haptoglobin antibody was purchased from DakoCytomation (Glostrup, Denmark). Endoprotease Glu-C (V8 protease) and Peptide-N4-(acetyl-β-D-glucosaminyl) asparagine amidase (PNGase F; E.C. 3.5.1.52, recombinant) were obtained from Roche Molecular Biochemicals (Tokyo, Japan). Alpha 1–3/4 fucosidase was purchased from Takara Bio Inc. (Shiga, Japan). Beta 1–4 galactosidase derived from Bacteroides fragilis and β 1–3 galactosidase derived from Xanthomonas manihotis was obtained from New England Biolabs Japan Inc. (Tokyo, Japan). Other reagents were of the highest quality or LC/MS grade available commercially.
Serum samples
Serum samples from normal volunteers (NV; n = 5; aged 40–70 years) and from patients with esophageal cancer (Eso; n = 5; 40–70 years; stage II or III; no metastasis and the tumor was operable), gastric cancer (Gas; n = 6; 40–70 years; stage II or III; no metastasis and the tumor was operable), colon cancer (Col; n = 18; 43–77 years; detailed information of patients shown in Supplementary Table 1), pancreatic cancer (Pan; n = 5; 40–70 years; stage II–IV; no metastasis and tumor was operable), gallbladder cancer (Gal; n = 6; 40–70 years; stage II or III; no metastasis and tumor was operable) and prostate cancer (Pro; n = 26; patients named “Pro 1–7” were 40–70 years, stage II or III, no metastasis and operable; patients named “Pro 8–26” were 53–83 years, stage I–III; detailed information for Pro 8–26 shown in Supplementary Table 2) were obtained from Osaka University-related Hospitals. Serum samples were stored at −80 °C until use.
Cell culture
An expression vector of human haptoglobin (pCDNA) was transfected into the human colon cancer cell line WiDr. Cells were grown in Dulbecco’s modified Eagle’s medium (Wako Pure Chemical Industries Ltd.) supplemented with 10 % fetal bovine serum (FBS) and 300 μg/ mL hygromycin (Sigma-Aldrich, St Louis, MO, USA) at 37 °C in an atmosphere of 5 % CO2. Single clones of WiDr cells expressing high levels of haptoglobin were used for subsequent experiments. When cell lines had reached 80 % confluence, each medium was replaced by antibiotic- and FBS-free medium after washing twice to remove FBS. Then, cell lines were incubated at 37 °C in an atmosphere of 5 % CO2 for 3 days, followed by collection of the conditioned media (which contained haptoglobin).
Purification of haptoglobin from human sera and cell cultured media
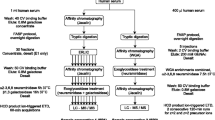
The procedure for purification of haptoglobin from sera was conducted as described in detail previously [6]. Briefly, the sera of patients with various types of cancer (100 μL) and NV (300 μL) were passed through a 0.45-μm cellulose acetate filter and diluted with buffer A (50 mM sodium phosphate buffer (pH 7.4), 0.5 M NaCl, 0.02 % NaN3) to a final volume of 7 mL. Diluted serum samples were passed five times through a human haptoglobin affinity column coupled with 300 μL of anti-human haptoglobin antibody at room temperature using a peristaltic pump. In the case of the culture medium, 250 mL of the medium containing haptoglobin was centrifuged at 220 g for 5 min and the supernatant filtered. The filtrated medium was circulated on the anti-human haptoglobin affinity column overnight at 4 °C. After washing the column with 15 mL of buffer A, followed by 5 mL of elution buffer (100 mM glycine, 0.5 M NaCl, pH 3.0), the haptoglobin bound to the column was eluted. The eluate was neutralized immediately with 100 μL of 2 M Tris–HCl (pH 8.0). The neutralized eluate containing haptoglobin was desalted using a PD-10 column (GE Healthcare, Piscataway, NJ, USA) equilibrated with water. One-twentieth of the desalted water containing haptoglobin derived from sera or the cell cultured medium was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 10 % polyacrylamide) under reduced conditions and then stained with Coomassie Brilliant Blue to confirm purification of haptoglobin. The remaining haptoglobin in the water was evaporated to dryness for subsequent analyses of glycopeptides and alditol N-glycans.
Preparation of desialo-glycopeptide of haptoglobin
The purified haptoglobin residue was dissolved in 500 μL of a reducing solution containing 250 mM Tris–HCl (pH 8.5), 6 M guanidine hydrochloride, 2 mM ethylenediamine tetra-acetic acid (EDTA) and 10 mg of dithiothreitol. The mixture was incubated at 50 °C for 1 h to reduce cysteine residues. After the addition of 20 mg of iodoacetamide to the mixture, the reaction was allowed to continue for 30 min at room temperature in the dark. The reaction mixture was pass through a Nap-5 column (GE Healthcare) equilibrated with water to remove salts from the reducing solution and excess iodoacetamide. The eluate containing S-carbamidomethylated haptoglobin (1 mL in water) was evaporated to dryness. The residue was dissolved with 100 μL of 50 mM NH4HCO3 containing an enzyme mixture of lysylendopeptidase (2 μg) and trypsin (2 μg) and incubated for 16 h at 37 °C. After boiling, the solution was mixed with endoprotease Glu-C (2 μg in 2 μL of 50 mM NH4HCO3). The mixture was incubated for 16 h at 37 °C and then boiled. To enrich glycopeptides, affinity separation by partitioning with Sepharose CL4B [35] was conducted. Briefly, water (100 μL) was added to the boiled solution, and the solution mixed with 1 mL of an organic solvent (1-butanol/ethanol; 4:1; v/v). The mixture was added to a 1.5-mL polypropylene tube containing a 100-μL packed volume of Sepharose CL4B equilibrated with 1-butanol/ethanol/water (4:1:1, v/v). After gentle agitation for 30 min, the gel was washed thrice with 1 mL of the same organic solvent. Then, the Sepharose CL4B gel was mixed gently with 400 μL of aqueous solvent, ethanol/water (1:1, v/v), and the liquid phase collected in the polypropylene tube. Four-hundred microliters of the same aqueous solvent was added to the gel again, and then shaken gently for 5 min. Then, the liquid phase was combined and evaporated to dryness. For desialylation, the residue was dissolved in 2 M acetic acid (200 μL) and incubated for 2 h at 80 °C. The solution was evaporated to dryness for LC-ESI MS analyses.
LC-ESI MS analyses of the desialo-glycopeptides of haptoglobin
Dried desialo-glycopeptides were dissolved in 20 μL of 0.08 % formic acid. Desialo-glycopeptides were separated using an ODS column (Develosil 300ODS-HG-5; 150 × 1.0 mm ID; Nomura Chemicals, Aichi, Japan) under specific gradient conditions. The mobile phases were solvent A (0.08 % formic acid) and solvent B (0.15 % formic acid in 80 % acetonitrile). The column was eluted with solvent A for 5 min, at which point the concentration of solvent B was increased to 40 % over 55 min at a flow rate of 50 μL/min using an Accela HPLC system (Thermo Fisher Scientific, Boston, MA, USA). The eluate was introduced continuously into an ESI source, and the glycopeptides were analyzed by LTQ Orbitrap XL (hybrid linear ion trap-orbitrap mass spectrometer; Thermo Fisher Scientific). In the MS setting, the voltage of the capillary source was set at 4.5 kV, and the temperature of the transfer capillary maintained at 300 °C. The capillary voltage and tube lens voltage were set at 15 and 50 V, respectively. MS data were obtained in positive ion mode over the mass range m/z 300 to m/z 3000 (resolution: 60000, mass accuracy: 5 ppm). MS/MS data were obtained by ion trap in LTQ Orbitrap XL (data dependent top 3, CID).
Relative quantitation of the glycoforms on each glycosylation site of haptoglobin
Quadruply charged ions [M+4H]4+ for site 1 or 4 and triply charged ions [M+3H]3+ for site 2 or 3 were selected for the relative quantitation of the glycoforms on each site. The peak intensity of highest isotope (not limited to monoisotope) of the corresponding glycoform in overall mass spectra was calculated using Xcalibur software ver. 2.0.7. (Thermo Fisher Scientific) by matching observed monoisotopic mass to theoretical monoisotopic mass (GlycoMod tool, http://web.expasy.org/glycomod/, mass tolerance; +/−0.01 Da) and also by confirming with MS/MS data (only for data dependent top 3 peaks). Although other charge state ions of the corresponding glycoforms were observed on each site, we chose quadruply charged ions for site 1 or 4 and triply charged ions for site 2 or 3, because their charge states provided the highest intensities of glycopeptide peaks on each site. Improving signal-to-noise ratio by using highest intensity charge state must lead to an increase in number of detectable minor components such as di-fucosylated glycopeptides.
Preparation of alditol -glycans of haptoglobin for analyses of fucosylation linkage
Purified haptoglobin from pooled sera was dissolved in 100 μL of water. This solution was dotted (2.5 μL × 4 times) onto a polyvinylidene difluoride (PVDF) membrane activated with ethanol. For haptoglobin derived from the WiDr medium, purified haptoglobin was dissolved in 10 μL of water, and then subjected to SDS-PAGE (10 % polyacrylamide) under reduced conditions. Proteins on one gel were transferred to a PVDF membrane under semi-dry conditions by means of a HorizeBLOT 2 M-R system (Atto Corp., Tokyo, Japan). The PVDF membrane was dried at room temperature overnight, washed with ethanol for 1 min, and then washed thrice for 1 min with water. To stain proteins, the membrane was incubated for 5 min with Direct Blue 71 (800 μL solution A: 0.1 % (w/v) Direct Blue 71 (Sigma–Aldrich) in 10 mL solution B :acetic acid:ethanol:water at 1:4:5). After destaining with solution B for 1 min, the PVDF membrane was dried at room temperature for >4 h. N-glycans were released from the dot-blotted or transferred proteins by the method of Wilson et al. [36] with some modifications by Nakano et al. [37]. The spots/bands of haptoglobin stained blue were cut out and placed in the separate wells of a 96-well microtiter plate. Then, the spots/bands were blocked with 100 μL of 1 % (w/v) polyvinylpyrrolidone 40,000 in 50 % (v/v) methanol, agitated for 20 min, and washed with water (100 μL × 5 times). PNGase F (2U in 10 μL of 100 mM phosphate buffer (pH 7.3), 25 mM of EDTA) was added to each well and preincubated at 37 °C for 15 min. Then, 10 μL of water was added to each well and incubated at 37 °C overnight to release N-glycans. During incubation, the 96-well plate was sealed with amplification tape to prevent evaporation. To collect released N-glycans, the plate was sonicated for 10 min and the solution containing released N-glycans (20 μL) transferred to 1.5-mL polypropylene tubes. The sample well was washed with water (50 μL × 2) and the washings combined. To transform the reducing end 1-amino N-acetylglucosamine to an N-acetylglucosamine after PNGase F release, ammonium acetate buffer (100 mM, pH 5.0, 20 μL) was added to the released N-glycans solution for 1 h at room temperature. After evaporation to dryness, the glycans were dissolved with 20 μL of 1 M NaBH4 in 50 mM KOH and reduced at 50 °C for 3 h to convert into N-glycan alditols. One microliter of acetic acid was added to stop the reaction, and N-glycan alditol solution was desalted using a cation-exchange column (35 μL). The column was washed twice with 50 μL of water to pass through the N-glycan alditols completely. After drying the desalted N-glycan alditol solution, borate contained in the sample was removed by the addition of 100 μL methanol and dried under a vacuum thrice. To remove sialic acid, the residue was dissolved in 2 M acetic acid (200 μL) and incubated for 2 h at 80 °C. The solution was evaporated to dryness.
LC-ESI MS analyses of the N-glycan alditols of haptoglobin
Dried N-glycan alditols were resuspended in 10 mM NH4HCO3 (15 μL) immediately before LC-ESI MS analyses. N-glycan alditols were separated using a porous graphitized carbon column (5 μm HyperCarb, 100 × 1.0 mm ID, Thermo Fisher Scientific) under specific gradient conditions. Separation of N-glycans was achieved using a sequence of isocratic and two segmented linear gradients: 0–8 min, 10 mM NH4HCO3; 8–38 min, 6.75–15.75 % (v/v) CH3CN in 10 mM NH4HCO3; 38–73 min, 15.75–40.5 % (v/v) CH3CN in 10 mM NH4HCO3; and increasing to 81 % (v/v) CH3CN in 10 mM NH4HCO3 for 10 min and re-equilibration with 10 mM NH4HCO3 for 6 min. The HPLC flow rate through the column was 50 μL/min using an Accela HPLC system (Thermo Fisher Scientific). With regard to mass spectrometer (LTQ Orbitrap XL), the voltage of the capillary source was set at 4.5 kV, and the temperature of the transfer capillary was maintained at 300 °C. The capillary voltage and tube lens voltage were set at 18 and 110 V, respectively. MS spectra were obtained in the positive ion mode using Orbitrap (mass range m/z 500 to m/z 2500), and MS/MS spectra were obtained using Iontrap after collision-induced dissociation. Monoisotopic masses were assigned with possible monosaccharide compositions using the GlycoMod tool available on the ExPASy server (http://au.expasy.org/tools/glycomod; mass tolerance for precursor ions is ±0.02 Da) and the proposed glycan structures were further verified through annotation using a fragmentation mass matching approach based on the MS/MS data by Xcalibur software ver. 2.0.7. (Thermo Fisher Scientific).
Results
Glycosylation site-specific analyses of haptoglobin by LC-ESI MS
To investigate if characteristic N-glycan structures were present among samples of various types of cancer, site-specific analyses of haptoglobin N-glycans were carried out using serum samples from patients with five types of gastroenterological cancer (Eso, Gas, Col, Gal, Pan), a non-gastroenterological cancer (Pro) and NV. Haptoglobin purified from sera was digested with a combination of trypsin, lysylendopeptidase and endopeptidase Glu-C after reduction and alkylation. Glycopeptides were enriched and desialylated, and the desialylated glycopeptides analyzed by LC-ESI MS. In theory, the glycopeptides digested with these three proteases should be: Met179-Glu194 including one glycosylation site (site 1: Asn184); Asn203-Glu210 including one glycosylation site (site 2: Asn207); Asn211-Lys215 including one glycosylation site (site 3: Asn211); and Val236-Asp246 including one glycosylation site (site 4: Asn241). The representative mass chromatogram for these four glycopeptide clusters for each glycosylation site is shown in Fig. 1. Glycopeptides from these four clusters include various desialylated N-glyans shown in Fig. 2. The average mass spectra during 2.5–3.5 min for glycopeptide cluster of site 3 are shown in Fig. 3a. The abbreviations for N-glycan structures in the glycopeptides are summarized in Fig. 2. For example, the peptide containing the tri-antennary N-glycan with one Fuc residue is represented as “3-F”. The first numeral indicates the branch number (tri-antennary in this case) and “F” indicates one Fuc residue. “0” denotes the absence of Fuc. Six glycopeptides (2-0, 2-F, 3-0, 3-F, 4-0, 4-F) were detected as mainly triply charged ion–proton adducts at site 3 in samples of various types of cancer and NV samples. The peak intensity of highest isotope (not limited to monoisotope) of the corresponding glycoform for site 3 in overall mass spectra was identified and calculated according to procedure described in material and methods section and Fig. 2. The peak intensities were shown in Fig. 3b. Total peak intensity of glycoform was set to 100 % on each sample, and then the relative percentage of each glycoform was calculated and shown in Fig. 3c. In Fig. 3, at site 3, fucosylated N-glycans (3-F, 4-F) were increased in samples of all types of cancer compared with NV samples. At site 1 and site 4, glycopeptides were detected as mainly quadruply charged ion–proton adducts (data not shown). At site 2, glycopeptides were detected as mainly triply charged ion–proton adducts (data not shown). For the comparison with NV, the ratio of fucosylated N-glycans was calculated based on the non-fucosylated corresponding N-glycan peaks at each site for all samples. For example, in case of tetra-antennary N-glycan at site 3 for NV #1 sample shown in Fig. 3, the ratio of monofucosylated N-glycan (4-F) to the non-fucosylated N-glycan (4–0) is 320/4880 (=0.0656). The results obtained from all samples are summarized in Fig. 4 and Supplementary Figs 1–4. Fucosylated N-glycans tended to increase at all glycosylation sites in cancer samples. At site 1, highly branched N-glycans (especially tetra-antennary N-glycans) were barely observed compared with those at other sites. In contrast, highly branched N-glycans (3–0 and 4–0) were the main ones at site 3, so fucosylated N-glycans (3-F, 3-FF, 4-F and 4-FF) could increase at site 3. In Eso samples, 3-FF and 4-F at site 2 as well as 2-F and 3-F at site 4 were significantly increased compared to NV samples (p < 0.01). In Gas samples, 3-FF and 4-F at site 2 as well as 3-F and 4-F at site 3 were significantly increased compared with NV samples (p < 0.01). In Col samples, 3-FF and 4-FF at site 2, 3-F and 4-F at site 3 and 2-F and 4-F at site 4 were significantly increased compared with NV samples (p < 0.01). In Pan samples, 4-F at site 2 was significantly increased compared with NV samples (p < 0.01). In Gal samples, 4-F at site 3 was significantly increased compared with NV samples (p < 0.01). In Pro samples, 3-FF at site 2, 4-F at site 3 as well as 3-FF and 4-F at site 4 were significantly increased compared with NV samples (p < 0.01). Non-fucosylated bi-antennary N-glycans (2–0) were significantly increased at site 1 only in Gas samples. Tri- and tetra-antennary N-glycans with two Fuc (3-FF and 4-FF) were observed at sites 1, 2 and 4 only in cancer samples. These difucosylated N-glycans (3-FF and 4-FF) at site 3 could not be detected. The reason for non-detection of difucosylated N-glycans at site 3 was overlapping of small amounts of miss-cleavaged monosialylated site 3 glycopeptides. This was because glycopeptides including site 3 were eluted very early and not separated on the ODS column according to differences in glycan structure. At other sites, miss-cleavaged sialylated glycopeptides were not observed.

Base peak chromatogram (BPC) of haptoglobin sample (this is representative data for Col #12) Four desialo-glycopeptide peaks were observed at 2.99 (site 3: Asn211), 36.41 (site 4: Asn241), 37.18 (site 2: Asn207) and 38.08 min (site 1: Asn184)

Abbreviations, structures, theoretical mass and observed mass for desialo-glycopeptides and N-glycan alditols detected in this study Desialo-glycopeptides which include sites 1 or 4 are calculated as quadruply charged ions, and those which include site 2 or 3 are calculated as triply charged ions. N-glycan alditols are calculated as doubly charged ions. *Presence of these glycopeptides was indeterminable cause of overlapping of small amounts of miss-cleavaged monosialylated their glycopeptides

Relative amount of N-glycans in glycopeptide cluster for site 3 derived from normal volunteer sample and various cancer samples. a Average mass spectra during 2.5–3.5 min for site 3. b Highest isotope peak intensity of the corresponding N-glycan in overall mass spectra for site 3. c Relative percentage of N-glycan at sites 3 after setting total peak intensity shown in (b) to 100 % on each sample. In fig. (a), table (b) and graph (c), NV (normal volunteer) #1, Eso (esophageal cancer) #2, Gas (gastric cancer) #4, Col (colon cancer) #9, Pan, (pancreatic cancer) #5, Gal (gallbladder cancer) #7, and Pro (prostate cancer) #25 were used for representative data. Abbreviations for glycan structures are summarized in Fig. 2

Ratio of glycopeptide fucosylated-glycoform to non-fucosylated-glycoform at each glycosylation site Glycopeptides were derived from haptoglobin purified from the sera of patients with Eso (n = 5), Gas (n = 6), Col (n = 18), Pan (n = 5), Gal (n = 6), Pro (n = 26) and sera of NV (n = 5). This ratio was calculated based on dividing peak intensity of highest isotope peak of fucosylated N-glycan by that of the corresponding non-fucosylated N-glycan. The glycoform abbreviations in this figure are summarized in Fig. 2. To compare NV and various cancer samples, the unpaired Student’s t-test (two-tailed) was used. The annotations with a single asterisk denote p < 0.05, and double asterisks denote p < 0.01
These results showed that the frequency of fucosylation at each site was different among the different types of cancer, but a site and glycan structure that increased fucosylation specifically in a particular type of cancer was not observed.
Identification of the linkage of fucosylation
Fucosylated N-glycans were increased in cancer samples compared with NV samples. To ascertain if the linkage type of fucosylation in increased fucosylated N-glycans among cancer types was different, N-glycans were released from purified haptoglobin by PNGase F and were analyzed by LC-ESI MS as N-glycan alditols. The BPC of N-glycan alditols (Fig. 5a) showed different glycoforms with different peaks among NV samples and cancer samples. Robust peaks at 35, 40 and 41 min were due to 2-0, 3-0 and 4-0, respectively. Many types of fucosylated N-glycan alditols (which are Lewis fucosylated bi-, tri- and tetra-antennary glycans and core fucosylated bi-, tri and tetra-antennary glycans) were detected as weak peaks in the BPC (Fig. 5a). This BPC showed that the ratio of fucosylated N-glycans on haptoglobin was increased in each cancer type sample. The EIC at m/z 1077.90–1077.92 (Fig. 5b) represented [M+2H]2+ of the monofucosylated tri-antennary N-glycan alditol. It showed differences in the relative abundance of Lewis fucosylated tri-antennary N-glycan alditol (3-F(L), 37 min) and core fucosylated tri-antennary N-glycan alditol (3-F(C), 44 min) between NV and cancer samples. Linkage of fucosylation was confirmed by digestion of fucosylated N-glycan alditol with α1-3/4 fucosidase (Supplementary Fig. 5). The EIC in Fig. 5b showed that Lewis fucosylated N-glycans were more abundant than core fucosylated N-glycans in five types of gastroenterological cancers (Eso, Gas, Col, Pan, Gal). In contrast, in a non-gastroenterological cancer (Pro), core-fucosylated N-glycans were more abundant than Lewis fucosylated N-glycans, which was the same pattern as with NV samples (although total amount of fucosylated N-glycans was different between NV samples and Pro samples). The increase in Lewis fucosylated N-glycans in gastroenterological cancer samples was also observed in bi- and tetra-antennary N-glycans (Supplementary Fig. 6). In samples of the non-gastroenterological cancer (Pro), a relative increase in Lewis fucosylated N-glycans was not observed instead, a relative increase in core fucosylated N-glycans was noted. In conclusion, analyses of N-glycan alditols demonstrated that linkage of fucosylation of haptoglobin derived from sera of patients with prostate cancer was different from that of gastroenterological cancer.

LC-ESI MS analyses of N-glycan alditols released from haptoglobin of normal volunteer samples (NV), five types of non-metastatic gastroenterological cancer samples (Eso_pool, Gas_pool, Col_pool, Pan_pool, Gal_pool), non-metastatic non-gastroenterological cancer samples (Pro_#18) and metastatic non-gastroenterological (Pro-Meta_#14) a The BPC obtained from analyses of various cancer samples and normal volunteer samples. b The EIC at m/z 1077.90–1077.92 suggests monofucosylated tri-antennary N-glycan alditol obtained from analyses of various cancer samples and normal volunteer samples. Lewis-fucosylated di, tri and tetra-antennary N-glycan (2-F(L), 3-F(L) and 4-F(L)) and core-fucosylated di, tri and tetra-antennary N-glycan (2-F(C), 3-F(C) and 4-F(C)) were assigned from the results of digestion with α1-3/4 fucosidase shown in Sup. Fig. 5
Change in linkage type of fucosylation by cancer metastasis
Among non-metastatic cancer samples, only the Pro sample showed different fucosylation patterns, i.e., core fucosylated N-glycans were more abundant than Lewis fucosylated N-glycans (Fig. 5b). To investigate the influence of metastasis on linkage of fucosylation, we analyzed N-glycan alditols released from metastatic Pro samples. The EIC of fucosylated tri-antennary N-glycan alditol (m/z 1077.90–1077.92) is shown in the bottommost panel of Fig. 5. This EIC in Fig. 5b demonstrated that, in metastatic Pro samples, Lewis-fucosylated N-glycan rather than core-fucosylated N-glycans were in the majority. This trend was also observed in fucosylated tetra-antennary N-glycans (bottommost panel in Supplementary Fig. 6). These results suggested that cancer metastasis transformed the linkage type of fucose from core-fucosylation to Lewis-fucosylation in Pro samples.
Comparison of the glycan structure of haptoglobin derived from a colon cancer cell line and from the sera of patients with colon cancer
In our previous study, the mechanism underlying production of fucosylated haptoglobin in Pan was studied [5]. Expression of haptoglobin mRNA was observed in a small number of Pan cell lines, suggesting that each cancer cell produced fucosylated haptoglobin. To investigate this possibility, we stably transfected the haptoglobin gene to a human colon cancer cell line (WiDr) to construct WiDr cell-expressing haptoglobin. After WiDr cells were cultured, haptoglobin was purified from the conditioned medium. Purified haptoglobin from WiDr cultured media (Cell-Hpt) and sera of patients with Col (Serum-Hpt) were separated from contaminant proteins by SDS-PAGE and transferred to PVDF membranes. N-glycans were released from haptoglobin on PVDF membranes by PNGase F and analyzed by LC-ESI MS as N-glycan alditols. In the BPC in Fig. 6a, Cell-Hpt samples and Serum-Hpt samples showed identical elution patterns with the same peaks of N-glycan structures. Comparison of the ratio of core- and Lewis-fucosylated N-glycans of Cell-Hpt with that of Serum-Hpt, the EIC of fucosylated tri-antennary N-glycans (m/z 1077.90–1077.92) is presented in Fig. 6b. This EIC showed that the ratio of core- and Lewis-fucosylated N-glycans were approximately identical. In fucosylated bi-antennary and tetra-antennary N-glycans, the ratio of core- and Lewis-fucosylated N-glycan was similar between Cell-Hpt and Serum-Hpt (Supplementary Fig. 7).

LC-ESI MS analyses of N-glycan alditols released from haptoglobin in the sera of patients with colon cancer (Serum-Hpt) and haptoglobin produced by a human colon carcinoma cell line (Cell-Hpt) a The BPC obtained from analyses of N-glycan alditols derived from purified haptoglobin from serum (Serum-Hpt) or cultured media (Cell-Hpt). b The EIC at m/z 1077.90–1077.92 indicate a monofucosylated tri-antennary N-glycan alditol obtained from analyses of Serum-Hpt and Cell-Hpt
These results demonstrated that the ratio of core-fucosylation and Lewis-fucosylation was not noticeably different between haptoglobin derived from a Col cell line and from the sera of patients with Col, suggesting that serum fucosylated haptoglobin in patients with colon cancer could be produced from colon cancer cells.
Discussion
Since we reported that fucosylated haptoglobin might be a novel tumor marker [5, 6], several researchers have studied fucosylated N-glycans on haptoglobin. However, information regarding comparable fucosylated N-glycan structures among various types of cancer has not been obtained because each research team used different methods to analyze N-glycans on haptoglobin. Here, we analyzed N-glycans on haptoglobin in the sera of patients with five types of gastroenterological cancers (Eso, Gas, Col, Gal and Pan), a non-gastroenterological cancer (Pro) and normal controls using the same analytical method to ascertain if characteristic fucosylation is observed in each type of cancer. Site-specific analyses revealed fucosylated N-glycans to be increased in cancer samples compared with NV samples, but characterizing the type of cancer by fucosylation linkage or fucosylation site of N-glycans on haptoglobin was difficult. Next, we analyzed the N-glycan alditols released from haptoglobin to identify the linkage of fucosylation. Lewis-fucosylated N-glycans were abundant in gastroenterological cancers (Eso, Gas, Col, Gal and Pan), whereas core-fucosylated N-glycans were abundant in prostate cancer.
Our previous study [5] suggested two mechanisms underlying the production of fucosylated haptoglobin: (i) each cancer cell and (ii) the liver (which then secreted it into blood). If almost all fucosylated haptoglobin in the sera of patients is derived from cancer cells, distinct differences in N-glycan structure should be observed because the glycosyltransferase expression involved in branch formation (and fucosylation) is different in each organ/tissue. However, remarkable differences in the structure or fucosylation of N-glycan were not detected in five types of gastroenterological cancers. This result suggested that almost all of the fucosylated haptoglobin in the sera of cancer patients might be produced in the liver. The fucosylated haptoglobin derived from cancer cells might be too minor a component to change the trends of glycosylation (including fucosylation).
In a healthy liver, core-fucosylated proteins produced from normal hepatocytes go to the apical side and are secreted into the bile duct [38]. In a micro-metastasized liver, core-fucosylated proteins secreted into blood cause destruction of the cellular polarity of hepatocytes [38]. It is well known that gastroenterological cancers tend to metastasize to the liver. We used non-metastatic gastroenterological cancer samples in the present study. Therefore, core-fucosylated N-glycans in sera were probably not increased. Instead, Lewis-fucosylated N-glycans were increased in the sera of patients with non-metastatic gastroenterological cancers. The reason for this increase in Lewis-fucosylated N-glycans was probably an increase in α1-3 fucosyltransferases such as FUT4 and FUT6 in the liver. An alternate reason could be an increase in secretion of Lewis-fucosylated N-glycans into blood from gastroenterological cancer cells due to FUT3-, FUT4- and FUT6-catalyzed α1-3/4 fucosylation [39, 40]. In our next study, we wish to confirm that core-fucosylated N-glycans on haptoglobin are increased in the sera of patients with metastatic gastroenterological cancers.
In contrast to gastroenterological cancers, it is well known that Pro cancer tends to metastasize to the bone and not to the liver. Therefore, the cellular polarity of hepatocytes is not destroyed in subjects with metastatic Pro cancer. In the present study, increases in core-fucosylated N-glycans in the sera of patients with metastatic Pro cancer was not observed instead, a decrease of core-fucosylated N-glycans was noted. This result suggests that core-fucosylated haptoglobin in the sera of patients with Pro cancer was produced mainly in the liver. However, we observed that the mRNA of haptoglobin and FUT8 (catalyzed α1-6 fucosyltransferase) was expressed in a Pro cancer cell line [41]. Therefore, a portion of haptoglobin containing core-fucosylated N-glycans may be produced in Pro cancer cells. When Pro cancer cells metastasized, the ratio of Lewis-fucosylation and core-fucosylation of haptoglobin in the sera of patients was inverted. In general, Lewis-fucosylation is very important for cancer cells to metastasize to another tissue because sialyl Lewis fucose is a ligand for selectin (a key molecule for metastasis). Some research teams have reported that expression of α1-3 fucosyltransferases such as FUT6 and FUT7 in Pro cancer cells support metastasis to bone [42, 43]. Therefore, haptoglobin produced from metastatic Pro cancer cells might be highly Lewis-fucosylated.
The present study showed that the simultaneous and comparative analyses of N-glycans on haptoglobin in the sera of patients with various types of cancer were useful to find differential markers of cancers. Significant differences in the structure of haptoglobin N-glycan was not detected among gastroenterological cancers, which enabled the differential diagnosis among gastroenterological cancers, in this study. However, the remarkable difference in the linkage of fucosylation in haptoglobin N-glycan was detected between gastroenterological cancers and non-gastroenterological cancer (Pro). Moreover, we found the linkage of fucosylation in haptoglobin N-glycan was different between localized and metastatic Prostate cancer samples.
Although detail mechanisms for fucosylation changes of haptoglobin remain unknown, it is important to know which cells produce fucosylated haptoglobin in patients with prostate cancer with/without metastasis. Further study is required to identify fucosylated haptoglobin immunohistochemically.
Abbreviations
- LC-ESI MS:
-
Liquid chromatography-electrospray ionization-mass spectrometry
- Hpt:
-
Haptoglobin
- NV:
-
Normal volunteers
- Eso:
-
Esophageal cancer
- Gas:
-
Gastric cancer
- Col:
-
Colon cancer
- Pan:
-
Pancreatic cancer
- Gal:
-
Gallbladder cancer
- Pro:
-
Prostate cancer
References
Becker, D.J., Lowe, J.B.: Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003)
Hakomori, S.: Aberrant glycosylation in tumors and tumor-associated carbohydrate antigen. Adv. Cancer Res. 52, 257–331 (1989)
Miyoshi, E., Moriwaki, K., Nakagawa, T.: Biological function of fucosylation in cancer biology. J. Biochem. 143, 725–729 (2008)
Dempsey, E., Rudd, P.M.: Acute phase glycoproteins: bystanders or participants in carcinogenesis? Ann. N. Y. Acad. Sci. 1253, 122–132 (2012)
Okuyama, N., Ide, Y., Nakano, M., Nakagawa, T., Yamanaka, K., Moriwaki, K., Murata, K., Ohigashi, H., Yokoyama, S., Eguchi, H., Ishikawa, O., Ito, T., Kato, M., Kasahara, S., Gu, J., Taniguchi, N., Miyoshi, E.: Fucosylated haptoglobin is a novel marker for pancreatic cancer: a detailed analysis of the oligosaccharide structure and a possible mechanism for fucosylation. Int. J. Cancer 118, 2803–2808 (2006)
Nakano, M., Nakagawa, T., Ito, T., Kitada, T., Hijioka, T., Kasahara, A., Tajiri, M., Wada, Y., Taniguchi, N., Miyoshi, E.: Site-specific analysis of N-glycans on haptoglobin in sera of patients with pancreatic cancer: a novel approach for the development of tumor markers. Int. J. Cancer 122, 2301–2309 (2008)
Black, J.A., Chan, G.F., Hew, C.L., Dixon, G.H.: Gene action in the human haptoglobins. III. Isolation of the α-chains as single gene products. Isolation, molecular weight, and amino acid composition of α and β chains. Can. J. Biochem. 48, 123–132 (1970)
Kurosky, A., Barnett, D.R., Lee, T.H., Touchstone, B., Hay, R.E., Arnott, M.S., Bowman, B.H., Fitch, W.M.: Covalent structure of human haptoglobin: a serine protease homolog. Proc. Natl. Acad. Sci. U. S. A. 77, 3388–3392 (1980)
Ferens-Sieczkowska, M., Olczak, M.: Carbohydrate structures of haptoglobin in sera of healthy people and a patient with congenital disorder of glycosylation. Z. Naturforsch. C 56, 122–131 (2001)
He, Z., Aristoteli, L.P., Kritharides, L., Garner, B.: HPLC analysis of discrete haptoglobin isoform N-linked oligosaccharides following 2D-PAGE isolation. Biochem. Biophys. Res. Commun. 343, 496–503 (2006)
Noda, K., Miyoshi, E., Gu, J., Gao, C.X., Nakahara, S., Kitada, T., Honke, K., Suzuki, K., Yoshihara, H., Yoshikawa, K., Kawano, K., Tonetti, M., Kasahara, A., Hori, M., Hayashi, N., Taniguchi, N.: Relationship between elevated FX expression and increased production of GDP-L-fucose, a common donor substrate for fucosylation in human hepatocellular carcinoma and hepatoma cell lines. Cancer Res. 63, 6282–6289 (2003)
Ang, I.L., Poon, T.C., Lai, P.B., Chan, A.T., Ngai, S.M., Hui, A.Y., Johnson, P.J., Sung, J.J.: Study of serum haptoglobin and its glycoforms in the diagnosis of hepatocellular carcinoma: a glycoproteomic approach. J. Proteome Res. 5, 2691–700 (2006)
Zhang, S., Shu, H., Luo, K., Kang, X., Zhang, Y., Lu, H., Liu, Y.: N-linked glycan changes of serum haptoglobin β chain in liver disease patients. Mol. Biosyst. 7, 1621–1628 (2011)
Zhang, S., Jiang, K., Sun, C., Lu, H., Liu, Y.: Quantitative analysis of site-specific N-glycans on sera haptoglobin β chain in liver diseases. Acta Biochim Biophys Sin (Shanghai) 45, 1021–1029 (2013)
Zhu, J., Lin, Z., Wu, J., Yin, H., Dai, J., Feng, Z., Marrero, J., Lubman, D.M.: Analysis of serum haptoglobin fucosylation in hepatocellular carcinoma and liver cirrhosis of different etiologies. J. Proteome Res. 13, 2986–2997 (2014)
Hoagland, L.F., Campa, M.J., Gottlin, E.B., Herndon, J.E., Patz, E.F.: Haptoglobin and posttranslational glycan-modified derivatives as serum biomarkers for the diagnosis of nonsmall cell lung cancer. Cancer 110, 2260–2268 (2007)
Arnold, J.N., Saldova, R., Hamid, U.M., Rudd, P.M.: Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics 8, 3284–3293 (2008)
Tsai, H.Y., Boonyapranai, K., Sriyam, S., Yu, C.J., Wu, S.W., Khoo, K.H., Phutrakul, S., Chen, S.T.: Glycoproteomics analysis to identify a glycoform on haptoglobin associated with lung cancer. Proteomics 11, 2162–2170 (2011)
Váradi, C., Mittermayr, S., Szekrényes, Á., Kádas, J., Takacs, L., Kurucz, I., Guttman, A.: Analysis of haptoglobin N-glycome alterations in inflammatory and malignant lung diseases by capillary electrophoresis. Electrophoresis 34, 2287–2294 (2013)
Zhao, J., Patwa, T.H., Qiu, W., Shedden, K., Hinderer, R., Misek, D.E., Anderson, M.A., Simeone, D.M., Lubman, D.M.: Glycoprotein microarrays with multi-lectin detection: unique lectin binding patterns as a tool for classifying normal, chronic pancreatitis and pancreatic cancer sera. J. Proteome Res. 6, 1864–61874 (2007)
Matsumoto, H., Shinzaki, S., Narisada, M., Kawamoto, S., Kuwamoto, K., Moriwaki, K., Kanke, F., Satomura, S., Kumada, T., Miyoshi, E.: Clinical application of a lectin-antibody ELISA to measure fucosylated haptoglobin in sera of patients with pancreatic cancer. Clin. Chem. Lab. Med. 48, 505–512 (2010)
Lin, Z., Simeone, D.M., Anderson, M.A., Brand, R.E., Xie, X., Shedden, K.A., Ruffin, M.T., Lubman, D.M.: Mass spectrometric assay for analysis of haptoglobin fucosylation in pancreatic cancer. J. Proteome Res. 10, 2602–2611 (2011)
Sarrats, A., Saldova, R., Pla, E., Fort, E., Harvey, D.J., Struwe, W.B., de Llorens, R., Rudd, P.M., Peracaula, R.: Glycosylation of liver acute-phase proteins in pancreatic cancer and chronic pancreatitis. Proteomics Clin. Appl. 4, 432–448 (2010)
Park, S.Y., Lee, S.H., Kawasaki, N., Itoh, S., Kang, K., Hee Ryu, S., Hashii, N., Kim, J.M., Kim, J.Y., Hoe Kim, J.: α1-3/4 fucosylation at Asn 241 of β-haptoglobin is a novel marker for colon cancer: a combinatorial approach for development of glycan biomarkers. Int. J. Cancer 130, 2366–2376 (2012)
Takeda, Y., Shinzaki, S., Okudo, K., Moriwaki, K., Murata, K., Miyoshi, E.: Fucosylated haptoglobin is a novel type of cancer biomarker linked to the prognosis after an operation in colorectal cancer. Cancer 118, 3036–3043 (2012)
Bones, J., Byrne, J.C., O’Donoghue, N., McManus, C., Scaife, C., Boissin, H., Nastase, A., Rudd, P.M.: Glycomic and glycoproteomic analysis of serum from patients with stomach cancer reveals potential markers arising from host defense response mechanisms. J. Proteome Res. 10, 1246–1265 (2011)
Thompson, S., Dargan, E., Turner, G.A.: Increased fucosylation and other carbohydrate changes in haptoglobin in ovarian cancer. Cancer Lett. 66, 43–48 (1992)
Turner, G.A., Goodarzi, M.T., Thompson, S.: Glycosylation of alpha-1-proteinase inhibitor and haptoglobin in ovarian cancer: evidence for two different mechanisms. Glycoconj. J. 12, 211–218 (1995)
Saldova, R., Royle, L., Radcliffe, C.M., Abd Hamid, U.M., Evans, R., Arnold, J.N., Banks, R.E., Hutson, R., Harvey, D.J., Antrobus, R., Petrescu, S.M., Dwek, R.A., Rudd, P.M.: Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology 17, 1344–1356 (2007)
Fujimura, T., Shinohara, Y., Tissot, B., Pang, P.C., Kurogochi, M., Saito, S., Arai, Y., Sadilek, M., Murayama, K., Dell, A., Nishimura, S., Hakomori, S.: Glycosylation status of haptoglobin in sera of patients with prostate cancer vs. benign prostate disease or normal subjects. Int. J. Cancer 122, 39–49 (2008)
Yoon, S.J., Park, S.Y., Pang, P.C., Gallagher, J., Gottesman, J.E., Dell, A., Kim, J.H., Hakomori, S.: N-glycosylation status of beta-haptoglobin in sera of patients with prostate cancer vs. benign prostate diseases. Int. J. Oncol. 36, 193–203 (2010)
Kazuno, S., Fujimura, T., Arai, T., Ueno, T., Nagao, K., Fujime, M., Murayama, K.: Multi-sequential surface plasmon resonance analysis of haptoglobin-lectin complex in sera of patients with malignant and benign prostate diseases. Anal. Biochem. 419, 241–249 (2011)
Abbott, K.L., Aoki, K., Lim, J.M., Porterfield, M., Johnson, R., O’Regan, R.M., Wells, L., Tiemeyer, M., Pierce, M.: Targeted glycoproteomic identification of biomarkers for human breast carcinoma. J. Proteome Res. 7, 1470–1480 (2008)
Abd Hamid, U.M., Royle, L., Saldova, R., Radcliffe, C.M., Harvey, D.J., Storr, S.J., Pardo, M., Antrobus, R., Chapman, C.J., Zitzmann, N., Robertson, J.F., Dwek, R.A., Rudd, P.M.: A strategy to reveal potential glycan markers from serum glycoproteins associated with breast cancer progression. Glycobiology 18, 1105–1118 (2008)
Wada, Y., Tajiri, M., Yoshida, S.: Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Anal. Chem. 76, 6560–6565 (2004)
Wilson, N.L., Schulz, B.L., Karlsson, N.G., Packer, N.H.: Sequential analysis of N- and O-linked glycosylation of 2D-PAGE separated glycoproteins. J. Proteome Res. 1, 521–529 (2002)
Nakano, M., Saldanha, R., Göbel, A., Kavallaris, M., Packer, N.H.: Identification of glycan structure alterations on cell membrane proteins in desoxyepothilone B resistant leukemia cells. Mol. Cell. Proteomics (2011). doi:10.1074/mcp.M111.009001
Nakagawa, T., Uozumi, N., Nakano, M., Mizuno-Horikawa, Y., Okuyama, N., Taguchi, T., Gu, J., Kondo, A., Taniguchi, N., Miyoshi, E.: Fucosylation of N-glycans regulates the secretion of hepatic glycoproteins into bile ducts. J. Biol. Chem. 281, 29797–29806 (2006)
Kaneko, M., Kudo, T., Iwasaki, H., Ikehara, Y., Nishihara, S., Nakagawa, S., Sasaki, K., Shiina, T., Inoko, H., Saitou, N., Narimatsu, H.: Alpha1,3-fucosyltransferase IX (Fuc-TIX) is very highly conserved between human and mouse; molecular cloning, characterization and tissue distribution of human Fuc-TIX. FEBS Lett. 452, 237–242 (1999)
Comelli, E.M., Head, S.R., Gilmartin, T., Whisenant, T., Haslam, S.M., North, S.J., Wong, N.K., Kudo, T., Narimatsu, H., Esko, J.D., Drickamer, K., Dell, A., Paulson, J.C.: A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology 16, 117–131 (2006)
Fujita, K., Shimomura, M., Uemura, M., Nakata, W., Sato, M., Nagahara, A., Nakai, Y., Takamatsu, S., Miyoshi, E.: Serum fucosylated haptoglobin as a novel prognostic biomarker predicting high-gleason prostate cancer. Prostate 74, 1052–1058 (2014)
Barthel, S.R., Wiese, G.K., Cho, J., Opperman, M.J., Hays, D.L., Siddiqui, J., Pienta, K.J., Furie, B., Dimitroff, C.J.: Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc. Natl. Acad. Sci. U. S. A. 106, 19491–19496 (2009)
Li, J., Guillebon, A.D., Hsu, J.W., Barthel, S.R., Dimitroff, C.J., Lee, Y.F., King, M.R.: Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br. J. Cancer 109, 3014–3022 (2013)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 335 kb)
Rights and permissions
About this article

Cite this article
Takahashi, S., Sugiyama, T., Shimomura, M. et al. Site-specific and linkage analyses of fucosylated N-glycans on haptoglobin in sera of patients with various types of cancer: possible implication for the differential diagnosis of cancer. Glycoconj J 33, 471–482 (2016). https://doi.org/10.1007/s10719-016-9653-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-016-9653-7