
Abstract
Genetic diversity of a set of 71 wheat accessions, including 53 biotype 2 Russian wheat aphid (RWA2)-resistant landraces and 18 RWA2 susceptible accessions, was assessed by examining molecular variation at multiple microsatellite (SSR) loci. Fifty-one wheat SSR primer pairs were used, 81 SSR loci were determined, and 545 SSR alleles were detected. These SSR loci covered all the three genomes, 21 chromosomes, and at least 41 of the 42 chromosome arms. Diversity values averaged over SSR loci were high with mean number of SSR alleles/locus = 6.7, mean Shannon’s index (H) = 1.291, and mean Nei’s gene diversity (He) = 0.609. The three wheat genomes ranked as A > D > B and the homoeologous groups ranked as 7 > 3 > 1 > 2 > 6 > 5 > 4 based on the number of alleles per locus. Xgwm136 on chromosome arm 1AS is the most polymorphic SSR locus with the largest number of observed and effective alleles and the highest H and He. Among all 2485 pairs of wheat accessions, genetic distance (GD) ranged from 0.054 to 1.933 and averaged 0.9832. A dendrogram based on GD matrix showed that all the wheat accessions could be grouped into distinct clusters. Most of the susceptible cultivars (13/18) were clustered into groups that contains all or mostly susceptible accessions. Most of the U.S. cultivars belong to a group that is distinguishable from all the different RWA2 resistant groups. Diversity analysis was also conducted separately for subgroups containing 53 RWA2-resistant accessions and 18 RWA2-susceptible accessions. Association mapping revealed 28 SSR loci significantly associated with leaf chlorosis, and 8 with leaf rolling. New chromosome regions associated with RWA2 resistance were detected, and indicated existence of new RWA resistance genes located on chromosomes of all other homoeologous groups in addition to the groups 1 and 7 in bread wheat. This information is helpful for development of mapping populations for RWA2 resistance genes from different phylogenetic groups, and for wise utilization of the RWA-resistant germplasm in wheat breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Russian Wheat Aphid (RWA), Diuraphis noxia (Kurdjumov), is an important insect pest of bread wheat, Triticum aestivum L., in several production areas of the world. The RWA was first discovered in the United States in 1986 (Webster et al. 1987) and has caused extensive damage to wheat production. Yield losses and increased production costs associated with RWA infestations were estimated to be >$800 million in the first 10 years after its introduction (Morrison and Peairs 1998) and additional losses have occurred since then (Peairs FB personal communication, Dep. Bioagricul. Sci. & Pest Mgmt., Colorado State Univ.). Control of RWA in the U.S. has critically relied on insecticides and, since 1996, on host-plant resistance genes effective against biotype 1 RWA (Smith et al. 2004). Most RWA-resistant wheat cultivars planted commercially in the United States contain the Dn4 gene from PI 372129 (Collins et al. 2005a). However, the emergence of a new biotype (designated as biotype 2) of RWA in 2003 poses a threat to existing wheat cultivars containing resistance genes against biotype 1, particularly Dn4 and Dny (Haley et al. 2004; Collins et al. 2005a; Jyoti and Michaud 2005; Qureshi et al. 2006).
Although the identification of biotype 2 represents the first detectable change in RWA populations in the United States, it is not necessarily the last (Qureshi et al. 2006). RWA populations from other parts of the world show considerable biotypic variation (Shufran et al. 1997; Basky 2003). Puterka et al. (1992) recognized at least eight RWA biotypes worldwide. In the United States, at least eight RWA biotypes were also recently identified, and biotype 2 is currently the predominant biotype (Peairs FB personal communication). Therefore, biotypic variation in RWA, whether introduced from exotic sources or evolved in situ, will likely continue to pose a serious threat to wheat production in the United States, mostly the west central Great Plains (Qureshi et al. 2006). Host-plant resistance is the most cost-effective and environmentally safe means for controlling RWA. Continuous efforts are necessary to identify and introduce additional resistance genes into commercially acceptable cultivars.
Among the 12 RWA resistance genes identified in wheat, rye (Secale cereale L.), and Aegilops tauschii (Smith et al. 2004; Peng et al. 2007), only the rye-derived Dn7 and its allelic Dn2414 are resistant to all the U.S. biotypes including biotype 2 (Anderson et al. 2003; Haley et al. 2004; Lapitan et al. 2007; Peng et al. 2007). The Dn7 gene has also been shown to provide effective protection from yield losses in field experiments (Collins et al. 2005b). However, Dn7 and its allelic Dn2414 are located on a 1RS.1BL wheat-rye translocation (Marais et al. 1994, 1998; Anderson et al. 2003; Lapitan et al. 2007; Peng et al. 2007) which is associated with poor bread-baking quality (Graybosch et al. 1990). In an effort to screen wheat germplasm accessions, Collins et al. (2005a) reported 58 (8.2%) of 709 accessions mainly from central Asia that showed resistance to RWA2. Some of these lines also confer resistance to RWA biotype 1 (http://www.ars-grin.gov/npgs/acc/acc_queries.html). These lines are mainly unimproved landraces of common wheat and thus do not likely contain the Secalin genes responsible for the poor bread-baking quality of wheat. To support breeders in selecting parental materials for breeding RWA resistance, knowledge of molecular genetic diversity, phylogenetic relationship of these resistant germplasm resources, and correlation between resistance traits and molecular markers would be helpful.
Simple-Sequence Repeats (SSRs), also known as microsatellites, are a class of molecular markers based on repeats of short (2–6 bp) DNA sequences (Litt and Luty 1989). The high level of polymorphism, combined with a high interspersion rate, makes them an abundant source of genetic markers. The usefulness of SSRs as genetic markers in plants has been demonstrated for several species, including soybean (Akkaya et al. 1995), rice (Wu and Tanksley 1993), maize (Senior and Heun 1993), Arabidopsis (Bell and Ecker 1994), and wheat (Bryan et al. 1997; Röder et al. 1998; Song et al. 2002; Peng and Lapitan 2005). These studies indicated that SSRs in plants can be up to ten-fold more variable than other marker systems such as Restriction Fragment Length Polymorphisms (RFLPs). Furthermore, the efficiency of SSR markers was also demonstrated for hexaploid wheat, a self-pollinating species with a relatively low level of intraspecific polymorphism (Plaschke et al. 1995; Röder et al. 1995). In recent years, a large number of SSR markers have been developed and extensively utilized in genomic mapping and marker-assisted breeding (Bryan et al. 1997; Röder et al. 1998; Peng et al. 1999, 2000a, b, c, 2003, 2007; Liu et al. 2001; Song et al. 2002; Arzani et al. 2004; Somers et al. 2004; http://maswheat.ucdavis.edu/), population genetic analyses (Li et al. 2000a, b) and diversity/polymorphism evaluation of germplasm (Fahima et al. 1998, 2002; Huang et al. 2002; Alamerew et al. 2004; Bertin et al. 2004; Khlestkina et al. 2004a, b; You et al. 2004; Roussel et al. 2004, 2005; Teklu et al. 2006; Zhang et al. 2006; Liu et al. 2007) in wheat.
Association mapping (AM) is to detect correlations between genotypes and phenotypes in a sample of individuals on the basis of linkage disequilibrium (LD) (Zondervan and Cardon 2004). As compared to other experimental designs that require sampling within families, AM offers the important advantage of sampling unrelated individuals in the population in the study of genetics of complex traits (Risch 2000). Sampling unrelated genotypes presents several advantages for marker-assisted plant breeding (Jannink et al. 2001). First, the experimental population can be a representative sample of the population to which inference is desired. Second, AM is more efficient in the use of resources, e.g., several traits can be studied in the same population using the same genotypic data, a higher proportion of molecular markers are likely to be polymorphic, providing better genome coverage than any bi-parental map. Furthermore, multi-year and multi-location phenotypic data may be available at no additional cost in study of elite lines (Rafalski 2002). In wheat, Breseghello and Sorrells (2006) demonstrated that association mapping in elite germplasm can enhance the information from QTL studies toward the implementation of marker-assisted selection.
The main objectives of this study were to determine genetic diversity and phylogenetic relationship among a group of 71 wheat accessions including 53 RWA2–resistant and 18 RWA2-susceptible genotypes for development of mapping populations for RWA2-resistance genes from different phylogenetic groups and for wise utilization of the RWA-resistant germplasm in wheat breeding programs. In the present paper, we report the differentiation and estimation of genetic diversity revealed by SSR markers among different wheat accessions originating primarily from central Asia selected for their high resistance to RWA2, and also potential SSR markers associated with RWA2 resistance.
Materials and methods
Plant materials
Previous studies showed that most of the bread wheat accessions resistant to RWA2 were from central Asia (Collins et al. 2005a; http://www.ars-grin.gov/npgs/acc/acc_queries.html). In this study, a set of 71 wheat genotypes was used. This bread wheat germplasm collection represents mainly landraces from central Asia and cultivars from the USA originating from geographically different locations (Table 1). Among these are 53 RWA2-resistant wheat accessions including 51 central Asian genotypes (38 from Iran, 10 from Afghanistan, 2 from Kazakhstan and 1 from Tajikistan), one Egyptian cultivar, and one Bulgarian cultivar, and 18 RWA2-susceptible wheat accessions including 10 U.S. genotypes developed in Colorado, Kansas, Oklahoma and Texas, six central Asian landraces, one Chinese cultivar/genetic material and one landrace from Sweden. The seeds were obtained from the National Small Grains Collection (NSGC) of the National Plant Germplasm System, USDA-ARS in Aberdeen, Idaho. Table 1 lists the geographic origin and reaction to RWA2 of these materials.
DNA extraction
Fifteen seeds were germinated in 100 × 15 mm Petri dishes (Becton Dickinson and Company, Franklin Lakes, NJ 07417–1886, USA), and equal amount of tissues were collected from 10 seedlings for each of the selected wheat genotypes. The tissue was placed in a 2 ml Eppendorf tube, immediately frozen in liquid nitrogen, and stored in a −80°C freezer. Total genomic DNA was extracted according to a procedure modified from Edwards et al. (1991).
SSR genotyping
In order to have complete coverage of the wheat genome, 51 primer pairs, at least one for each chromosome arm, which amplify the expected wheat SSR fragments were chosen for the analysis. These SSR primers included 48 GWMs (Röder et al. 1998), one BARC (Song et al. 2002), one CFD (Guyomarc’h et al. 2002), and one CWEM (Peng and Lapitan 2005). Primer designation, the amplified loci, the chromosome arm location, the number of alleles, and the range of allele size are presented in Table 2. Primer sequences are available at the GrainGenes web site: http://wheat.pw.usda.gov/cgi-bin/graingenes/browse.cgi?class=marker.
The polymerase chain reactions (PCR) were performed in PTC-200 MJ Thermocyclers (MJ Research, Inc., Watertown, MA). The PCR procedure was the same as in Peng et al. (1999). The PCR-amplified fragments were separated by electrophoresis on a 5% denaturing polyacrylamide gel. The gels were visualized with silver staining (Bio-Rad Kit Protocol #LIT-34 89–0559 689; Morrissey 1981). As an example, Fig. 1 shows the polymorphism pattern of SSR marker Xgwm161.

Amplification profiles of SSR marker Xgwm161 in the 71 wheat accessions/cultivars. Lanes 1–71 corresponds to line ID in Table 1. M = 10 bp DNA ladder
Genetic data analysis
The SSR markers were treated as co-dominant markers. The visualized polyacrylamide gels were scored using capital alphabets following the user guide of Popgene (Yeh and Yang 2000). The collection of wheat germplasm was classified into three groups, R+S group including all the 71 accessions, R group consisting of 53 RWA2-resistant accessions, and S group consisting of 18 RWA2-susceptible accessions. These three groups were subjected to the following analyses. The actual number of alleles was counted for each amplified locus. The effective number of alleles was estimated as n = 1 + 4N e u for each locus, where N e is the effective population size and u is the average mutation rate (Kimura and Crow 1964). Shannon’s (1949) information index (H) was estimated for each locus using the formula H = −∑p i ln p i (i = 1 to S), where S is the total number of alleles in the locus, and p i is the proportion of S made up of the ith allele. Gene diversity was estimated according to the formula of Nei (1973) for each locus He = 1 − ∑P 2 ij , where P ij is the frequency of the jth allele for ith locus summed across all alleles of the locus. Anderson et al. (1993) referred to gene diversity as the Polymorphic Information Content (PIC). Nei’s (1972) genetic identity (I) and genetic distance (D) were calculated for each pair of tested entries according to the equations: I = J XY /√ J X J Y , and D = −ln I, where J X , J Y and J XY are the arithmetic means of j X (=∑x 2i ), j Y (=∑y 2i ) and j xy (=∑xiyi), respectively, over all loci; xi and yi are the frequencies of the ith allele in X and Y entry/population, respectively. The un-weighted pair-group method with arithmetic average (UPGMA) was chosen as a clustering method. Based on the Nei’s GD, the dendrogram was drawn using UPGMA. All these analyses were conducted using the Popgene 1.32 Window-based computer package (Yeh and Yang 2000).
Association mapping
Leaf chlorosis (LC) and leaf rolling (LR) were scaled to measure reaction of the bread wheat accessions to RWA2 as described by Collins et al. (2005a). Associations between these two traits and the available SSR data were analyzed based on the whole set of 71 bread wheat accessions used in the present study. The AM was conducted using the general linear model by the aid of the computer software TASSEL2.0.1 (http://www.maizegenetics.net/tassel) accounting for population structure estimates from STRUCTURE2.1 software (Prichard et al. 2000, http://pritch.bsd.uchicago.edu/structure.html). The number of permutation run was set as 10,000 to obtain the permutation-based test of marker significance and the experiment-wise P-value for marker significance. Markers were declared to be associated with a RWA2 resistance trait only when the markers are significant (P < 0.05) in all the three tests, F-test, permutation-based test, and experiment-wise test.
Results and discussion
Genome coverage of the SSR markers
In the present study, the 51 selected primer pairs amplified 81 SSR loci in the set of wheat germplasm collections used (Tables 1 and 2). Each of these SSR primer pairs amplified 1–5 loci. The chromosome arm locations for 67 of these SSR loci were known, and unknown for other 14 (Table 2). The 67 SSR loci were distributed on all but the 7DL chromosome arm of hexaploid wheat (Tables 2 and 3). Therefore, the 81 SSR loci covered all the three genomes, seven homoeologous groups, 21 chromosomes, and at least 41 of the 42 chromosome arms of wheat. In previous studies of genetic diversity in wheat germplasm, scientists used from 20 to 30 SSRs (Fahima et al. 1998, 2002; Huang et al. 2002; Alamerew et al. 2004; Khlestkina et al. 2004a, b; Bertin et al. 2004; Teklu et al. 2006; Zhang et al. 2006; Liu et al. 2007), and a few studies used around 40 SSRs (Roussel et al. 2004, 2005). Thus, the genetic information revealed in this study represents the wheat genome with higher coverage.
SSR polymorphism and genetic diversity
High levels of polymorphism were observed for the SSR markers. A total of 545 alleles were detected on 81 SSR loci in a set (R+S) of 71 wheat collections including both the RWA2-resistant and -susceptible lines, with a range of 2–24 and average of 6.7 alleles per SSR locus. The most polymorphic SSR locus was Xgwm136 on chromosome arm 1AS which had the largest number of observed (24) and effective (13.6) alleles, and the highest Shannon’s index and Nei’s gene diversity (Table 2). Among the three genomes of hexaploid wheat, the A genome had the highest number of SSR alleles per locus (8.05), followed by D (7.70) and B (6.54) genomes. Homoeologous group 7 had the highest number of alleles per locus (11.25), followed by group 3 (9.50), group 1 (7.57), group 2 (7.22), and other groups (< 7.0) (Table 3).
For the R and S subgroups of the wheat accessions, the total number of detected SSR alleles was 494 and 362, respectively, and the average number of SSR alleles was 6.1 and 4.5, respectively (Table 2). In comparison with the R+S group, the R subgroup had slightly lower total and average numbers of alleles, but the S subgroup had a obviously lower total and average numbers of detected alleles (Table 2). Thus, high levels of polymorphism still occur in the RWA2-resistant germplasm, but relatively lower polymorphism exists in the RWA2-susceptible accessions. The origin and geographic distribution of the wheat germplasm showed in Table 1 may explain this result.
The number of alleles per SSR locus is one of the most important parameters describing polymorphism and varies from 4.6 to 18.2 in previous studies of wheat genetic diversity (Fahima et al. 1998, 2002; Huang et al. 2002; Bertin et al. 2004; Khlestkina et al. 2004b; Roussel et al. 2004, 2005; You et al. 2004; Teklu et al. 2006; Liu et al. 2007). Many of these wheat diversity studies treated a SSR marker as a single locus. However, studies have shown that many wheat SSR markers could actually determine multiple loci (Röder et al. 1998; Peng et al. 2000c; Sourdille et al. 2004). We strictly differentiated the SSR loci from markers in this study. Therefore, the previous studies usually reported higher number of alleles per locus than we did in the present study (Table 2).
An allele found in only one accession is termed an accession-specific allele. A total of 104 SSR alleles were found to be accession-specific, and amounted to 19.08% of the total number of alleles (Table 2). In spelt wheat, Bertin et al. (2004) found that 17% of the SSR alleles were accession specific. This indicates that some aspect of SSR polymorphism in the analyzed bread wheat germplasm is slightly higher than that in spelt wheat (19% vs. 17%). Chromosome locations are known for 97 of the 104 accession-specific alleles (Table 2). These SSR alleles were randomly distributed among the three genomes, i.e., 37, 33, and 27 on A, B, and D genomes, respectively (χ2 = 1.57, P = 0.4566). However, distribution of accession-specific alleles was not random among homoeologous groups (χ2 = 24.70, P = 0.0004), i.e., 29 on group 1, 12 on each of groups 2 and 3, 8 on group 4, 6 on group 5, 13 on group 6 and 17 on 7. Thus, homoeologous groups 1 and 7 showed higher numbers of accession-specific SSR alleles (Table 3). Out of the 71 wheat accessions, 14 showed three or more accession-specific alleles. Chinese Spring originally from China, PI 135064 from Afghan, and PI 243659 from Iran, showed 7, 4, and 4 accession-specific alleles, respectively (Table 2).
If all the alleles in an SSR locus were equally frequent, the proportion (F ≈ 1/(1 + 4N e u)) of homozygotes would be the reciprocal of the number of alleles at the locus maintained in the population. Therefore, n = 1/F may be used as a measure of the effective number of alleles maintained in the population, which in general will be less than the actual number (Kimura and Crow 1964). The effective number of alleles ranged from 1.121 to 13.585 with an average of 3.71 for the 81 SSR loci detected in the R+S group (Table 2). This effective number of alleles is positively correlated with the observed number of alleles (r = 0.93, P < 0.001). This means that the proportion of homozygotes would decrease, or heterozygosity/polymorphism would increase, with increase of the actual number of alleles at a locus in the analyzed bread wheat germplasm.
In comparison with the R+S group, both the R and S subgroups had lower total and average numbers of effective alleles estimated. For the R+S group, and R and S subgroups, the total number of effective alleles was 300.2, 279.7, and 259.9, respectively, and the average number of effective alleles was 3.71, 3.45, and 3.21, respectively (Table 2). However, this difference of number of effective alleles among the three groups of wheat accessions seems not as significant as that for the observed number of alleles.
Diversity indices provide important information about rarity and commonness of alleles at a locus. The Shannon diversity index (H) is one common diversity index often used to characterize allele diversity in a locus. Shannon’s index accounts for both abundance and evenness of the alleles present (Shannon and Weaver 1949), and is useful for understanding allele structure at an SSR locus. Among the 81 SSR loci in the R+S group of wheat accessions, H averaged 1.291 and ranged from 0.249 to 2.866. For those loci with H > 2, the number of alleles must be ≥12, and for those loci with H < 1, the number of alleles would be ≤4 (Table 2). Thus, H is positively correlated with the allele number (r = 0.94, P < 0.001), and can be used to quantify the diversity or polymorphism of SSR markers. In 15 wild emmer wheat (T. dicoccoides) populations representing a wide range of ecological conditions of soil, temperature, and water availability in Israel and Turkey, this parameter (H) averaged 0.84 and ranged 0.166–1.307 (Fahima et al. 2002), and was lower than those (mean = 1.291, 1.226 and 1.134 for R+S, R, and S group or subgroup, respectively) reported in the present study (Table 2). Therefore, there is high genetic diversity as revealed by SSR markers in the bread wheat germplasm analyzed, and this diversity is even higher than that in the wild emmer wheat, the progenitor of cultivated tetraploid durum and hexaploid bread wheats. The genetic diversity of wheat thus have not decayed but increased during the long process of domestication of wild progenitor and cultivation/spread from Middle East to other parts of the world. This may be because of the narrowing of the wheat germplasm base can be averted and the genetic diversity can be subsequently increased through the introgression of novel materials, as Reif et al. (2005) reported.
From the R+S to R, and S group or subgroup, H decreased from 1.291 to 1.226, and to 1.134 (Table 2). There was little difference (0.065) between R+S group and R subgroup for H. This difference of H between R+S group and S subgroup was much larger (0.157), and also large (0.092) between R and S subgroups. This indicates again that there exists high genetic diversity in the RWA2-resistant wheat germplasm.
Nei’s (1973) gene diversity or expected heterozygosity (He) is another common diversity index in population genetics and is equivalent to PIC. In the present study, gene diversity varied from 0.108 to 0.926 and averaged 0.609. He was >0.9 for SSR loci Xgwm136, Xgwm88a, Xgwm111c, Xgwm146, Xgwm314, and Xgwm372. Each of these loci had an allele number >15 (Table 2). The correlation between gene diversity and allele number is positive and highly significant (r = 0.79, P < 0.001). Bertin et al. (2004) found a value of He equal to 0.78 (0.323–0.936), analyzing spelt wheat accessions. Fahima et al. (2002) reported a He values of 0.5 (0.094–0.736) in analyzing wild emmer wheat accessions. Huang et al. (2002) showed a He value of 0.77 (0.43–0.94) in analysis of a set of common wheat germplasm from across all wheat producing regions. Roussel et al. (2004) revealed 0.662 (0.214–0.868) of He value in evaluating French bread wheat accessions. Roussel et al. (2005) also found a He value of 0.650 (0.211–0.899) in testing Eurasian bread wheat varieties. Khlestkina et al. (2004b) obtained a value of He equal to 0.70 (0.46–0.82) in evaluating Siberian common spring wheat. Teklu et al. (2006) analyzed Ethiopian tetraploid wheat landraces and found a He value equal to 0.684–0.688. Liu et al. (2007) showed a He value of 0.56 (0.18–0.80) in analyzing a Chinese wheat gene pool from recurrent selection. In the present study, He was estimated as 0.609, 0.591, and 0.581 for R+S, R, and S group or subgroup, respectively (Table 2). There was minor He difference (0.010–0.018) among these three group/subgroups of wheat accessions. Therefore, gene diversity of RWA2-resistant subgroup of wheat accessions is similar to that of the whole group involving resistant and susceptible accessions. Gene diversity as reflected by He index in the present study (Table 2) is comparable with previously published results of different wheat species or populations, but the range (0.818 = 0.926–0.108 for R+S, 0.846 = 0.919–0.073 for R subgroup, and 0.883 = 0.883–0.000 for S subgroup) is the greatest. This indicates that gene diversity is highly variable among different SSR loci in the bread wheat germplasm used in the present study.
The observed heterozygosity (He-a), proportion of observed heterozygotes at a given locus, was estimated for each of the 81 loci. The result showed that heterozygosity occurred only on five loci, accounting for 6.17% of the total SSR loci investigated. Furthermore, these five loci showed heterozygosity only distributed in 1AL, 3AL, 6DL, 7BS and an unknown chromosome regions (Table 2). It seems that heterozygote is a rare event detected only by a few SSR loci on a few chromosome regions in the bread wheat germplasm resistant or susceptible to RWA2. The average heterozygosity for the R+S, R, and S group or subgroups was 0.016, 0.018, and 0.012, respectively. This low rate of observed heterozygosity is expected because wheat is a typical self-pollinated crop species with outcrossing rates <1% and more than 1% for wheat plants grown in close proximity (Waines and Hegde 2003), and may explain why the observed heterozygosity has been simply ignored in the previous studies in wheat germplasm (Fahima et al. 1998, 2002; Huang et al. 2002; Alamerew at al 2004; Khlestkina et al. 2004a, b; Bertin et al. 2004; Roussel et al. 2004, 2005; Teklu et al. 2006; Zhang et al. 2006; Liu et al. 2007).
Contribution of A, B, D genomes to the genetic variation of wheat
Among the 67 SSR loci of known chromosome locations, 19 were located in A, 28 in B, and 20 in D genome, and the detected allele numbers were 153, 183, and 154 for A, B, and D genome, respectively (Table 3). The average number of alleles per locus for A, B, and D genome was 8.05, 6.54, and 7.70, respectively. This means that the alleles/locus of A genome is 34.6% and 4.5% higher than B and D genome, respectively; and D genome is 17.7% higher than B genome. Therefore, for the set of wheat accessions including both the RWA2-resistant and -susceptible used in this study, the contribution of A, B, and D genome to the genetic variation revealed by SSR markers can be ranked as A > D > B. But the three genomes can be ranked as D (1.470) > A (1.280) > B (1.264) based on Shannon index (H), and D (0.667) > B (0.613) > A (0.610) based on Nei’s gene diversity (He) (Table 3). Roussel et al. (2004) also reported the inconsistent ranking based on allele number/locus and gene diversity. In a group of stripe rust-resistant T. dicoccoides accessions, the A genome possessed 20% higher number of SSR alleles than the B genome (Fahima et al. 1998). In an analysis of quantitative trait locus (QTL) in T. dicoccoides, the number of domestication-related QTL effects and domestication syndrome factors in the A genome was found to be higher than in the B genome due to the higher polymorphism for expressed traits in the A genome (Peng et al. 2003). In a set of 998 bread wheat accessions from 68 countries worldwide, the three genomes were ranked as B > A > D for both the SSR allele number and gene diversity (Huang et al. 2002). In a set of 96 random accessions of Chinese bread wheat, the three genomes were also ranked as B > A > D for SSR allele number (You et al. 2004). In French bread wheat accessions, the three genomes were ranked as A > D > B based on SSR alleles/locus and B > D > A based on gene diversity or PIC value (Roussel et al. 2004). In Ethiopian hexaploid wheat, the three genomes were ranked as B > A > D based on SSR alleles per locus (Alamerew et al. 2004). In Siberian common spring wheat, more SSR alleles were also detected in B genome than in A and D genomes (Khlestkina et al. 2004b). The A genome was more polymorphic than the B genome in the three Ethiopian tetraploid wheat species, T. durum, T. dicoccon, and T. turgidum (Teklu et al. 2006). In general, the A genome is more polymorphic than B or D genome in tetraploid wheat and some hexaploid wheat materials/ populations, and B genome is more polymorphic than A or D genome in many of the hexaploid wheat accessions.
Genetic distance
With the aid of Popgene computer program (Yeh and Yang 2000), Nei’s (1972) GD and Nei’s (1978) unbiased GD were estimated for the 71 × (71 − 1)/2 = 2485 possible pairs/combinations of wheat accessions. The results indicated that the GD is quite similar to the unbiased genetic distance in the present study. The GD ranged from 0.054 to 1.933 with an average of 0.9832 (Appendix 1). The wheat collections used in this study have a wide geographical distribution and are mainly landraces. Large GD is thus expected in this set of wheat collections. The two Iranian landraces PI 621458 and PI 621462, both collected from East Azerbaijan, have the closest GD (0.054). The largest GD (1.933) occurred between Colorado breeding line CO970547-7, susceptible to RWA2, and Egyptian cultivar Bouhi 12 (PI 366103), resistant to RWA2. Thus a cross between CO970547-7 and Bouhi 12 could be used to develop a mapping population for the RWA2 resistance gene carried by Bouhi 12 because high SSR mapping efficiency would be expected in this cross.
The SSR-based GD is 0.69 (0.018–0.964) among a set of stripe rust-resistant T. dicoccoides accessions (Fahima et al. 1998), 1.862 (0.876–3.320) among 15 T. dicoccoides populations (Fahima et al. 2002), 0.30 (0.08–0.71) in set of Siberian common spring wheat varieties (Khlestkina et al. 2004b), 0.26 between T. durum and T. turgidum, 0.38 between T. turgidum and T. dicoccon, 0.34 between T. durum and T. dicoccon (Teklu et al. 2006). The GD among the bread wheat accessions (R+S group) in the present study is smaller than that among T. dicoccoides populations, but larger than that between T. dicoccoides accessions, Siberian common spring wheat cultivars, and even tetraploid wheat species. This again indicates a high genetic polymorphism existing in the bread wheat accessions used in this study.
We also estimated separately the pair-wise GD for R and S group of wheat accessions (data not shown) using the same set of 81 SSR loci. The results showed that the GD estimated in R or S subgroup was the same as that obtained in the R+S group for a specific pair of accessions. This is because of the large GD (Appendix 1) and the low observed heterozygosity (Table 2) in the wheat accessions used in the present study. The number of individuals to be used for estimating GD can be very small if the genetic distance is large and the average heterozygosity of the two species compared is low (Nei 1978).
Phylogenetic analysis
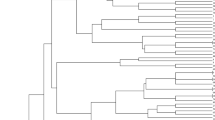
Based on Nei’s original GD (Appendix 1), cluster analysis was carried out using the UPGMA method and resulted in a phylogenetic dendrogram shown in Fig. 2. The 71 wheat accessions could be divided into two mega-groups. Mega-group I included 20 wheat accessions that can be further clustered into four subgroups and mega-group II contained 51 accessions that could be clustered into nine subgroups. The pattern of clustering for most of the wheat accessions corresponded with the geographic distribution of wheat collections. This result is in agreement with several previous studies (Fahima et al. 1998, 2002; Huang et al. 2002; Bertin et al. 2004; You et al. 2004; Roussel et al. 2005).

Dendrogram of 71 wheat genotypes based on the Nei’s (1972) original genetic distance calculated from data of 81 SSR loci, using the UPGMA as the clustering method. Marked groups are described in the section of result and discussion
Subgroup Ia contained only two accessions with relatively large GD (0.722), the genetically well characterized cultivar Chinese Spring, and a Colorado modern cultivar Yuma, both of which are susceptible to RWA2. Subgroup Ib contained five RWA2-resistant Iranian landraces. Subgroup Ic contained eight RWA2-susceptible modern cultivars developed and/or deployed in the western Great Plains of USA. Subgroup Id contained five central Asian accessions (one from Tajikistan, two from Kazakhstan, and one from each of Afghanistan and Iran), one of which was RWA2-susceptible (Fig. 2).
Subgroup IIa consisted of 18 accessions of which the majority (16) were RWA2-resistant Iranian landraces, one was a RWA2-resistant Egyptian cultivar, and one was a RWA2-susceptible Azerbaijan landrace. Subgroup IIb consisted of four accessions including one RWA2-resistant Bulgarian cultivar CItr 11349 (Varna 20), one RWA2-resistant Iranian landrace, one RWA2-resistant Afghanistan landrace, and one RWA2-susceptible Iranian landrace. Subgroup IIc contained one RWA2-susceptible Turkmenistan cultivar and two RWA2-resistant Iranian landraces. Subgroup IId contained four accessions including only one RWA2-resistant Iranian landrace and three RWA2-susceptible accessions (one Sweden landrace, one Afghanistan landrace and one US breeding line). Subgroup IIe contained seven RWA2-resistant Afghanistan landraces or cultivated materials and one RWA2-susceptible Iranian landrace. Subgroup IIf consisted of only two RWA2-resistant landraces, one from each of Iran and Afghan. Subgroup IIg consisted of eight RWA2-resistant landraces of which seven from Iran and one from Afghan. Subgroup IIh contained two RWA2-resistant Iranian landraces with GD of 0.587. Subgroup IIi consisted of two genetically distant RWA2-resistant Iranian landraces with GD of 0.919 (Fig. 2, Appendix 1).
As indicated in Fig. 2, among the 18 RWA2-susceptible accessions, 11 (61%) belonged to the mega-group I and the other seven (39%) belonged to mega-group II. Further analysis indicated that 13 (72%) of the 18 susceptible accessions were assigned to subgroups Ia, Ic and IId; 100% of the accessions in subgroups Ia and Ic, and 75% of the accessions in subgroup IId were susceptible to RWA2. Among the 53 RWA2-resistant accessions, 9 (17%) belonged to the mega-group I, and the other 44 (83%) belonged to the mega-group II. These resistant accessions distributed among >10 subgroups, indicating the existence of a rich genetic diversity among these bread wheat germplasm.
Phylogenetic analyses were also conducted for the R and S subgroup of the wheat accessions. The resulting dendrograms were presented in Figs. 3 and 4. It was indicated that the 53 RWA2-resistant wheat accessions could still be divided into two mega groups and further into many distinguishable subgroups (Fig. 3), and the classification was similar to that of whole set of accessions shown in Fig. 2. For example, all the resistant accessions in mega-group I (Fig. 2) were still in the same mega-group as showed in Fig. 3 with one exception of PI 621256 that belonged to mega-group II in analysis of the R+S group. In this analysis of R subgroup, majority (43) of the 53 RWA2-resistant accessions were still classified into the mega-group II, but each of the subgroups IIa and IIe in Fig. 2 were further divided into two subgroups in Fig. 3. In the analysis of S subgroup including 18 RWA2-susceptible accessions, the 11 accessions belonging to mega-group I and the 7 accessions belonging to mega-group II in Fig. 4 maintained basically the same classification as shown in Fig. 2. The 7 RWA2-susceptible accessions classified into five subgroups in Fig. 2 could be re-classified into two phylogenetic subgroups (Fig. 4).


This phylogenetic information of the relatedness of RWA2-resistant wheat accessions is of value for wheat breeding programs worldwide. The RWA2-susceptible USA cultivars belonged to a separate phylogenetic mega-group or subgroups from that of RWA2-resistant accessions originating from central Asia. Thus, the crosses between the susceptible U.S./Colorado cultivars and the RWA2-resistant central Asian landraces or cultivars could introduce RWA resistance genes into Colorado wheat cultivars and meanwhile enhance the genetic diversity that is essential for sustainability of wheat cultivars. The phylogenetic grouping shown in Figs. 2 and 3 can be utilized to diversify the source of resistance genes against RWA2, and perhaps increase the chance of diversity in response to new biotypes of RWA. It is likely that individuals in one cluster carry genes that are different from individuals in another cluster. However, this hypothesis needs to be tested using gene mapping approaches and further allelism tests for those located in the same chromosome regions.
In an attempt to estimate the different contributions of each genome to the genetic variation of wheat, GDs were calculated based on each genome set of SSR loci separately. The resulting dendrograms (Appendix 2–4) showed that the clustering patterns are different from each other among A, B, and D genome, and from the whole wheat genome shown in Fig. 2. However, the grouping of RWA2-susceptible U.S. cultivars was similar for the genome subset of SSR loci and the whole genome, and particularly the A genome that had grouping pattern most similar to that based on the whole genome.
Association between RWA resistance and SSR markers
LC and LR are the two important traits used to determine reaction of wheat plants to infestation of the Russian wheat aphid (Anderson et al. 2003; Collins et al. 2005a, 2005b; Peng et al. 2007). AM could detect association between phenotype and genotype based on LD (Zondervan and Cardon 2004). In the present study, we used the AM approach to find SSR markers and chromosome regions potentially associated or linked with RWA2 resistance traits. The results showed that there were as many as 28 SSR marker loci showed significant correlation with LC (p-M < 0.01, p-P < 0.01, p-adj < 0.05). Xgwm427b showed marginally significant association with LC (p-M < 0.01, p-P < 0.01, p-adj = 0.0596). These marker loci could explain 10.49–40.80% of the total variation after fitting the other model effects (Table 4). However, there were only eight SSR marker loci showed significant association with LR (p-M < 0.01, p-P < 0.01, p-adj < 0.05). Xgwm369 showed marginally significant association with LR (p-M < 0.01, p-P < 0.01, p-adj = 0.0549). These marker loci could explain 10.77–36.86% of the total variation (Table 4). According to Breseghello and Sorrells (2006), before marker-assisted selection based on the markers identified via AM can be applied to the progeny, a simple and essential step of confirmation is required for individual cultivars involved in crosses. This confirmation is necessary because the marker alleles are correlated with, but not entirely predictive of, the gene alleles. To confirm the association with the marker locus, for example, we can genotype F2 plants and phenotype F3 progeny as we did in wheat genome mapping (Peng et al. 1999, 2000a, b, 2003, 2007; Lapitan et al. 2007).
Up to date, 12 resistance genes against RWA have been identified, and most of them are located on group 1 and group 7 chromosomes of Triticeae (1D, 1R, and 7D) (Smith et al. 2004; Peng et al. 2007). In the present study, we found, with the aid of AM method, 29 SSR loci significantly associated with RWA resistance reflected by LC and/or LR in a set of bread wheat germplasm including both the RWA2-resistant and -susceptible accessions. Theses loci distributed on at least 16 chromosomes and 21 chromosome arms. Except for the groups 1 and 7 chromosomes harboring the currently known RWA resistance genes, other 11 chromosomes belonging to 2, 3, 4, 5 and 6 homoeologous groups were also found to be associated with RWA2 resistance, especially LC (Table 4). Nevertheless, the number of chromosomes and homoeologous groups associated with LR was obviously smaller than those with LC, and additional homoeologous groups were 3, 4, and 6 except for the group 1 and 7. LC and LR are important traits reflecting RWA resistance but are not necessarily controlled by the same genes. Many loci associated with LC may not be correlated with LR. But all the loci for LR showed significant associations with LC (Table 4). Thus many genetic loci control LC, and part (1/3 ∼ 9/29) of them control both LR and LC. The loci associated with RWA2 resistance on homoeologous groups 2, 3, 4, 5, and 6 must be new genes that have not been mapped, yet.
Abbreviations
- AM:
-
Association mapping
- LC:
-
Leaf chlorosis
- LD:
-
Linkage disequilibrium
- LR:
-
Leaf rolling
- GD:
-
Genetic distance
- PCR:
-
Polymerase chain reaction
- PIC:
-
Polymorphic information content
- RWA:
-
Russian wheat aphid
- SSR:
-
Simple sequence repeat
- UPGMA:
-
The un-weighted pair-group method with arithmetic average
References
Akkaya MS, Shoemaker RC, Specht JE, Bhagwat AA, Cregan PB (1995) Integration of simple-sequence repeat DNA markers into a soybean linkage map. Crop Sci 35:1439–1445
Alamerew S, Chebotar S, Huang XQ, Röder MS, Börner A (2004) Genetic diversity in Ethiopian hexaploid and tetraploid wheat germplasm assessed by microsatellite markers. Genet Resour Crop Evol 51:559–567
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186
Anderson GR, Papa D, Peng JH, Tahir M, Lapitan NLV (2003) Genetic mapping of Dn7, a rye gene conferring resistance to the Russian wheat aphid in wheat. Theor Appl Genet 107:1297–1303
Arzani A, Peng JH, Lapitan NLV (2004) DNA and morphological markers for a Russian wheat aphid resistance gene. Euphytica 139:167–172
Basky Z (2003) Biotypic and pest status differences between Hungarian and South African populations of Russian wheat aphid, Diuraphis noxia (Kurdjumov) (Homoptera: Aphididae). Pest Manag Sci 59:1152–1158
Bell CJ, Ecker JR (1994) Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics 19:137–144
Bertin P, Grégoire D, Massart S, de Froidmont D (2004) High level of genetic diversity among spelt germplasm revealed by microsatellite markers. Genome 47:1043–1052
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Bryan GJ, Collins AJ, Stephenson P, Orry A, Smith JB, and Gale MD (1997) Isolation and characterization of microsatellites from hexaploid bread wheat. Theor Appl Genet 94:557–563
Collins MB, Haley SD, Peairs FB, Rudolph JB (2005a) Biotype 2 Russian wheat aphid resistance among wheat germplasm accessions. Crop Sci 45:1877–1880
Collins MB, Haley SD, Randolph TL, Peairs FB, Rudolph JB (2005b) Comparison of Dn4- and Dn7-carrying spring wheat genotypes artificially infested with Russian wheat aphid (Homoptera: Aphididae) biotype 1. J Econ Entol 98:1698–1703
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucl Acids Res 19:1349
Fahima T, Röder MS, Grama A, Nevo E (1998) Microsatellite DNA polymorphism divergence in Triticum dicoccoides accessions highly resistant to yellow rust. Theor Appl Genet 96:187–195
Fahima T, Röder MS, Wendehake K, Kirzhner VM, Nevo E (2002) Microsatellite polymorphism in natural populations of wild emmer wheat (Triricum dicoccoides) in Israel. Theor Appl Genet 104:17–29
Graybosch RA, Peterson CJ, Hansen LW, Mattern PJ (1990) Relationships between protein solubility characteristics, 1BL/1RS, high molecular weight glutenin composition, and end-use quality in winter wheat germplasm. Cereal Chem 67:342–349
Guyomarc’h H, Sourdille P, Charmet G, Edwards K, Bernard M (2002) Characterization of polymorphic microsatellite markers from Aegilops tauschii and transferability to the D-genome of bread wheat. Theor Appl Genet 104:1164–1172
Haley SD, Peairs FB, Walker CB, Rudolph JB, Randolph TL (2004) Occurrence of a new Russian wheat aphid biotype in Colorado. Crop Sci 44:1589–1592
Huang XQ, Börner A, Röder MS, Ganal MW (2002) Assessing genetic diversity of wheat (Triticum aestivum L.) germplasm using microsatellite markers. Theor Appl Genet 105:699–707
Jannink JL, Bink M, Jansen RA (2001) Using complex plant pedigrees to map valuable genes. Trends Plant Sci 6:337–342
Jyoti JL, Michaud JP (2005) Comparative biology of a novel strain of Russian wheat aphid (Homoptera: Aphididae) on three wheat varieties. J Econ Entomol 98:1032–1039
Khlestkina EK, Huang XQ, Quenum FJB, Chebotar S. Röder MS, Börner A (2004a) Genetic diversity in cultivated plants—loss or stability? Theor Appl Genet 108:1466–1472
Khlestkina EK, Röder MS, Efremova TT, Börner A, Shumny VK (2004b) The genetic diversity of old and modern Siberian varieties of common spring wheat as determined by microsatellite markers. Plant Breed 123:122–127
Kimura M, Crow JF (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Lapitan NLV, Peng JH, Sharma V (2007) A high-density map and PCR markers for Russian wheat aphid resistance gene Dn7 on chromosome 1RS/1BL. Crop Sci 47:809–818
Li YC, Röder MS, Fahima T, Kirzhner VM, Beiles A, Korol AB, Nevo E (2000a) Natural selection causing microsatellite divergence in wild emmer wheat at the ecologically variable microsite at Ammiad, Israel. Theor Appl Genet 100:985–999
Li YC, Fahima T, Peng JH, Röder MS, Kirzhner VM, Beiles A, Korol AB, Nevo E (2000b) Edaphic microsatellite DNA divergence in wild emmer wheat, Triticum dicoccoides, at a microsite: Tabigha, Israel. Theor Appl Genet 101:1029–1038
Litt M, Luty JA (1989) A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiacmuscle actin gene. Am J Hum Genet 44:397–401
Liu XM, Smith CM, Gill BS, Tolmay V (2001) Microsatellite markers linked to six Russian wheat aphid resistance genes in wheat. Theor Appl Genet 102:504–510
Liu J, Liu L, Hou N, Zhang A, Liu C (2007) Genetic diversity of wheat gene pool of recurrent selection assessed by microsatellite markers and morphological traits. Euphytica 155:249–258
Marais GF, Horn M, Du Toit F (1994) Intergeneric transfer (rye to wheat) of a gene(s) for Russian wheat aphid resistance. Plant Breed 113:265–271
Marais GF, Wessels WG, Horn M (1998) Association of a stem rust resistance gene (Sr45) and two Russian wheat aphid resistance genes (Dn5 and Dn7) with mapped structural loci in common wheat. S Af J Plant Soil 15:67–71
Morrissey JH (1981) Silver stain for protein in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal Biochem 117:307–310
Morrison WP, Peairs FB (1998) Response model concept and economic impact. In: Quisenberry SS, Peairs FB (eds) A response model for an introduced pest—The Russian wheat aphid Thomas Say Publ in Entomology, Entomology Soc Am. Lanham, MD, pp 1–11
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Peng JH, Lapitan NLV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomics 5:80–96
Peng JH., Fahima T, Röder MS, Li YC, Dahan A, Grama A, Ronin YI, Korol AB, Nevo E (1999) Microsatellite tagging of stripe-rust resistance gene YrH52 derived from wild emmer wheat, Triticum dicoccoides, and suggestive negative crossover interference on chromosome 1B. Theor Appl Genet 98:862–872
Peng JH, Fahima T, Röder MS, Huang QY, Dahan A, Grama A, Nevo E (2000a) High-density molecular map of chromosome region harboring stripe-rust resistance gene YrH52 and Yr15 derived from wild emmer wheat, Triticum dicccoides. Genetica 109:199–210
Peng JH, Fahima T, Röder MS, Li YC, Grama A, Nevo E (2000b) Microsatellite high-density mapping of stripe-rust resistance gene YrH52 region on chromosome 1B and evaluation of its marker-assisted selection in F2 generation in wild emmer wheat. New Phytologist 146:141–154
Peng JH, Korol AB, Fahima T, Röder MS, Ronin YI, Li YC, Nevo E (2000c) Molecular genetic maps in wild emmer wheat, Triticum dicoccoides: Genome-wide coverage, massive negative interference, and putative quasi-linkage. Genome Res 10:1509–1531
Peng JH, Ronin YI, Fahima T, Röder MS, Li YC, Nevo E, Korol AB (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494
Peng JH, Wang H, Haley SD, Peairs FB, Lapitan NLV (2007) Molecular mapping of the Russian wheat aphid resistance gene Dn2414 in wheat. Crop Sci 47:2418–2429
Plaschke J, Ganal MW, Röder MS (1995) Detection of genetic diversity in closely related bread wheat using microsatellite markers. Theor Appl Genet 91:1001–1007
Prichard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–659
Puterka GJ, Burd JD, Burton RL (1992) Biotypic variation in a worldwide collection of Russian wheat aphid (Homoptera: Aphididae). J Econ Entomol 85:1497–1506
Qureshi JA, Michaud JP, Martin TJ (2006) Resistance to biotype 2 Russian wheat aphid (Homoptera: Aphididae) in two wheat lines. J Econ Entomol 99:544–550
Rafalski JA (2002) Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci 162:329–333
Reif JC, Zhang P, Dreisigacker S, Warburton ML, van Ginkel M, Hoisington D, Bohn M, Melchinger AE (2005) Wheat genetic diversity trends during domestication and breeding. Theor Appl Genet 110:859–864
Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856
Röder MS, Plaschke J, König SU, Börner A, Sorrells ME, Tanksley SD, Ganal MW (1995) Abundance, variability and chromosomal location of microsatellites in wheat. Mol Gen Genet 246:327–333
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Roussel V, Koenig J, Beckert M, Balfourier F (2004) Molecular diversity in French bread wheat accessions related to temporal trends and breeding programmes. Theor Appl Genet 108:920–930
Roussel V, Leisova L, Exbrayat F, Stehno Z, Balfourier F (2005) SSR allelic diversity changes in 480 European bread wheat varieties released from 1840 to 2000. Theor Appl Genet 111:162–170
Senior ML, Heun M (1993) Mapping maize microsatellites and polymerase chain reaction confirmation of the targeted repeats using a CT primer. Genome 36:884–889
Shannon CE, Weaver W (1949) The mathematical theory of communication. Univ of Illinois Press, Urbana
Shufran KA, Burd JD, Webster JA (1997) Biotypic status of Russian wheat aphid (Homoptera: Aphididae) in the United States. J Econ Entomol 90:1684–1689
Smith CM, Belay T, Stauffer C, Stary P, Kubeckova I, Starkey S (2004) Identification of Russian wheat aphid (Homoptera: Aphididae) populations virulent to the Dn4 resistance gene. J Econ Entomol 97:1112–1117
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L). Theor Appl Genet 109:1105–1114
Song QJ, Fickus EW, Cregan PB (2002) Characterization of trinucleotide SSR motifs in wheat. Theor Appl Genet 104:286–293
Sourdille P, Singh S, Cadalen T, Brown-Guedira GL, Gay G, Qi L, Gill BS, Dufour P, Murigneux A, Bernard M (2004) Microsatellite-based deletion bin system for the establishment of genetic–physical map relationships in wheat (Triticum aestivum L). Funct Integr Genom 4:12–25
Teklu Y, Hammer K, Huang XQ, Röder MS (2006) Analysis of microsatellite diversity in Ethiopian tetraploid wheat landraces. Genet Resour Crop Evol 53:1115–1126
Waines JG, Hegde SG (2003) Intra-specifi c gene flow in bread wheat as aff ected by reproductive biology and pollination ecology of wheat flowers. Crop Sci 43:451–463
Webster JA, Starks KJ, Burton RL (1987) Plant resistance studies with Diuraphis noxia(Homoptera: Aphididae) a new United States wheat pest. J Econ Entomol 80:944–949
Wu KS, Tanksley SD (1993) Abundance, polymorphism and genetic mapping of microsatellites in rice. Mol Gen Genet 241:225–235
Yeh FC, Yang RC (2000) POPGENE Version 1.32. University of Albert and Center for International Research, http://www.ualberta.ca/∼fyeh/
You GX, Zhang XY, Wang LF (2004) An estimation of the minimum number of SSR loci needed to reveal genetic relationships in wheat varieties: Information from 96 random accessions with maximized genetic diversity. Mol Breed 14:397–406
Zhang P, Dreisigacker S, Buerkert A, Alkhanjari S, Melchinger AE, Warburton ML (2006) Genetic diversity and relationships of wheat landraces from Oman investigated with SSR markers. Genet Resour Crop Evol 53:1351–1360
Zondervan KT, Cardon LR (2004) The complex interplay among factors that influence allelic association. Nat Rev Genet 5:89—100
Acknowledgements
This study was supported in part by the Chinese Academy of Sciences under the Important Directional Program of Knowledge Innovation Engineering Grant No. KSCX2-YW-Z-0722, the Colorado Wheat Research Foundation, the U.S. Department of Agriculture under Cooperative Agreements USDA Contract No. 2001–52100-11293 and USDA Contract No. 2003-34205-13636, the National Research Initiative of USDA’s Cooperative State Research, Education and Extension Service, CAP Grant No. 2006-55606-16629, and Hatch Funds Project No. 644.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1
Appendix 2

Appendix 3

Appendix 4

Rights and permissions
About this article
Cite this article
Peng, J.H., Bai, Y., Haley, S.D. et al. Microsatellite-based molecular diversity of bread wheat germplasm and association mapping of wheat resistance to the Russian wheat aphid. Genetica 135, 95–122 (2009). https://doi.org/10.1007/s10709-008-9262-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-008-9262-x