
Abstract
To assess the utility of the tomato fruit-specific E8 gene’s promoter for driving vaccine antigen expression in plant, the 2.2 kb and 1.1 kb E8 promoters were isolated and sequenced from Lycopersicon esculentum cv. Jinfeng #1. The 1.1 kb promoter was fused to vaccine antigen HBsAg M gene for the transfer to Nicotiana tabacum, and the CaMV 35S promoter was used for comparison. Cholera toxin B (ctb) gene under the control of the 1.1 kb promoter was transformed into both N. tabacum and L. esculentum. Southern blot hybridization confirmed the stable integration of the target genes into the tomato and tobacco genomes. ELISA assay showed that the expression product of HBsAg M gene under the control of the 1.1 kb E8 promoter could not be detected in transgenic tobacco tissues such as leaves, flowers, and seeds. In contrast, the expression of HBsAg M gene driven by CaMV 35S promoter could be detected in transgenic tobacco. ELISA assay for CTB proved that the 1.1 kb E8 promoter was able to direct the expression of exotic gene in ripe fruits of transgenic tomato, but expression was absent in leaf, flower, and unripe fruit of tomato, and CTB protein was not detected in transgenic tobacco tissues such as leaves, flowers, and seeds when the gene was under the control of the 1.1 kb E8 promoter. The results indicated that the E8 promoter acted not only in an organ-specific, but also in a species-specific fashion in plant transformation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The developments in the plant transgenic techniques make the production of transgenic plant-made oral vaccines possible. Plant-produced proteins are considered to be safe, as plants do not harbor human pathogens such as human immunodeficiency virus, hepatitis viruses, and toxins, as animal/animal cell culture systems often do (Larrick and Thomas 2001). Several factors need to be considered in order to maximize the yield of recombinant proteins in transgenic plants, such as codon usage, stable and efficient expression of foreign gene, etc. However, the procedure, timing, and localization of gene expression are regulated by a hierarchy of control mechanisms. Promoter, as one of main transcript regulators, plays a significant role in gene expression.
The Cauliflower mosaic virus 35S (CaMV 35S) promoter can efficiently drive foreign gene expression in plant cells (Jani et al. 2002). However, this promoter does not confer any specificity-neither tissue specificity nor plant developmental stage specificity on exogenous gene expression leading to lower expression levels (Smigocki and Owens 1988). As for transgenic plant vaccines, low expression level will lead to the decrease of immunogenicity, which will trigger insufficient protection or immunological tolerance to human body.
A major advantage of targeting protein expression to fruits is that edible parts can be consumed uncooked or partially processed, making them convenient sites for the production of vaccines. Several fruit-specific promoters such as E4, E8, PG and 2A11 were identified in tomato (Coupe and Deikman 1997; Deikman et al. 1998). These promoters have been mostly used to investigate the role of ethylene in fruit ripening. The E8 promoter is one of the most extensively characterized ripening-specific tomato promoters. The deletion studies of flanking DNA sequences on E8 gene expression in transgenic tomato fruit showed that the E8 promoter has at least two main regions contributing to its transcriptional regulation: the region of −2181 to −1088 containing DNA sequence that confer ethylene responsiveness in unripe fruit but are sufficient for E8 gene expression during ripening, and the ‘downstream’ region of −1088 to the transcriptional start site is sufficient for ripening-specific transcription in the absence of ethylene synthesis (Deikman et al. 1992). Some researches used the E8 promoter to drive the expression of exogenous genes in transgenic tomato fruits (Sandhu et al. 2000; Krasnyanski et al. 2001; Mehta et al. 2002; Yakoby et al. 2006), however, there is no report on the gene expression driven by the E8 promoter in the non-tomato system as far as we know. In order to explore the gene expression profile of antigen protein gene(s) driven by E8 promoter and to compare the expression with other plant beside tomato, we expressed the hepatitis B virus middle Antigen (HBsAg M) gene and the cholera toxin B (ctb) gene driven by the 1.1 kb E8 promoter in tobacco and tomato. This work should be of interest to those using tissue-specific promoter such as E8 for expression of antigen genes in heterologous plants.
Materials and methods
Bio-materials and vectors
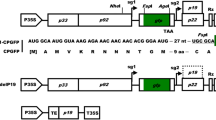
Escherichia coli Top10, Agrobacterium tumefaciens LBA4404 and plants tobacco (Nicotiana tobacum cv. Xanthi) kept by this laboratory and tomato (Lycopersicon esculentum cv. Suifeng and cv. Jinfeng #1) purchased from Guangzhou Institute of Vegetable Sciences were used in this study. Plant materials were cultured under a 16 h light/8 h dark photoperiod at 25°C on Murashige and Skoog medium (MS) supplemented with 0.7% agar and 3% sucrose. Plasmid pMD18-T (TaKaRa) was used as a TA-cloning vector to clone the products of polymerase chain reaction (PCR). Plasmid pBluescript II SK (Stratagene) was used as an intermediate vector for cloning and pBI121 was used to construct plant expression vectors pBIS2S, pBI-1.1E8-S2S and pJES1. In pBI-1.1E8-S2S, a DNA fragment containing the HBsAg M gene (Gan et al. 1987) under the control of the 1.1 kb E8 promoter was inserted between sites of Hind III and Sac I in pBI121. The construct pBIS2S, harbouring the CaMV 35S promoter to control the expression of the HBsAg M gene, was used as a reference of pBI-1.1E8-S2S. In pJES1, a DNA fragment containing the ctb gene under the control of the 1.1 kb E8 promoter was inserted between sites of Hind III and Sac I in pBI121 (Fig. 1).

Plant transformation vectors used in the experiments. NOSp: nopaline synthase promoter; NOSt: nopaline synthhase terminator; NPT II: neomycin phosphotransferase II gene conferring resistance to kanamycin and used as a selectable marker for plant transformation. 35S-p: cauliflower mosaic virus 35S promoter. 1.1 E8-p: 1.1-kb tomato fruit specific E8 promoter. HBV S2 + S: hepatitis B virus surface antigen middle protein gene (HBsAg M). ctb: cholera toxin B gene
Molecular manipulations and PCR primers
Plasmid extraction, enzyme digestion, ligation, and bacterial transformations were performed as described by Sambrook et al. (1992).
PCR primers used in this experiment were listed in Table 1. Primer pairs P1 and P2 for amplifying the 852-bp fragment encoding HBsAg M (preS2 + S) gene were designed according to Gan et al. (1987). Primer pairs P3 and P4 for amplifying the 375-bp fragment encoding the region of ctb gene were designed according to GenBank (accession number: U25679).
Two pairs of primers were designed to clone the 1.1 kb and 2.2 kb E8 promoters from genomic DNA extracted from L. esculentum cv. Jinfeng #1. Primer pair of P5 and P6 for 1.1 kb E8 promoter was based on the sequence described by Deikman and Fischer (1988) and primer pair of P7 and P6 for 2.2 kb E8 promoter was based on the sequence described by Deikman et al. (1992) and by Deikman and Fisher (1988).
PCR conditions are as follows: an initail denaturation at 94°C for 5 min; 35 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C for 1.5 min; followed by a 5 min incubation at 72°C. PCR products were subcloned into pMD18-T vector and sequenced by the Bioasia Biotechnology Company, Shanghai.
Plant transformation
Plant expression vectors were transformed into Agrobacterium tumefaciens LBA4404 by freeze-thaw method and confirmed by PCR. Agrobacterium clone containing plant expression construct was propagated under selection of kanamycin in YEB medium (5 g/l of each of tryptone, yeast extract, and sucrose, 2 mM MgSO4, pH 7.2) at 28°C and 250 rpm to OD 0.4–0.6 and was used to infect explants of tobacco or tomato.
For tobacco transformation, leaves of in-vitro-grown tobacco were cut into 0.5–1 cm2 sections and then immersed in the suspension of 109 Agrobacterium cells per ml for 30 s. The explants were then blotted dry on a sterilized filter paper and cultured on co-cultivation medium (MS + 6-benzylaminopurine (BA) 1 mg/l + α-naphthaleneacetic acid (NAA) 0.1 mg/l) for 48 h. After co-cultivation, the explants were transferred onto a selection medium (MS + BA 1 mg/l + NAA 0.1 mg/l + kanamycin 100 mg/l + cefotaxime 300 mg/l). All explants were transferred to fresh medium every 3 weeks. The developed shoots were transferred onto rooting medium (MS + kanamycin 100 mg/l + cefotaxime 100 mg/l), on which roots grew out in succession after culturing for one week. The plantlets with healthy roots were planted in a greenhouse.
For tomato transformation, cotyledons of in-vitro-germinated tomato seedlings of 10–14 days old were cut off from the third base position and inoculated on MS for 2 days. Thereafter, the induced cotyledon explants were dipped in the solution of 109 Agrobacterium cells per ml for 30 s, blotted dry on sterilized filter paper, and placed on co-cultivation medium (MS + BA 3 mg/l + indolacetic acid (IAA) 0.1 mg/l) for 48 h. After co-cultivation, the explants were transferred to selection medium (MS + BA 3 mg/l + IAA 0.1 mg/l + kanamycin 100 mg/l + cefotaxime 300 mg/l). The developed shoots on selection medium for 4–6 weeks were transferred onto elongation medium (MS + BA 2 mg/l + kanamycin 100 mg/l + cefotaxime 100 mg/l). When the shoots reached about 3 cm in height, they were then transferred into rooting medium (MS + IAA 0.2 mg/l + kanamycin 100 mg/l + cefotaxime 100 mg/l) for cultivation for 10 days, on which the roots were induced. The plantlets were transferred to MS plus kanamycin and cefotaxime for further growth. The plantlets of about 5–10 cm height and with healthy roots were planted in a greenhouse.
All plant cultures were kept at 25–28°C and under a 16/8 h (light/dark) photoperiod.
Detection of transgene in transformed plants
Total nucleic acids were extracted from 0.3–0.5 g of leaf tissue of transgenic and wild-type plants according to the cetyltrimethylammonium bromide (CTAB) protocol described by Doyle and Doyle (1990). PCR amplification was initially carried out to detect the presence of the target gene in transgenic tobacco and tomato. The reaction condition for amplification was as described above and the PCR results were then confirmed by Southern blot analysis. 10 μg of total plant genomic DNA digested with selected restriction enzymes was transferred to nylon membranes. Probes used were the PCR products amplified from either the plasmid pBI-1.1E8-S2S with primer pair P1 and P2 or from the plasmid pJES1 with primer pair P3 and P4. Preparation of probes and hybridization were performed by the use of the DIG-High Prime Labeling and Detection Kit (Roche).
Detection of targeting proteins in transgenic plants
Fresh leaf tissue (0.5 g) of transgenic tobacco and the wild-type plant were ground in liquid nitrogen with a mortar and pestle. The samples were collected and mixed with 0.5 ml of PBS (0.8% NaCl, 0.02% KCl, 10 mM sodium phosphate, pH 7.4), and placed at 4°C overnight (Thanavala et al. 1995). The mixture was then centrifuged at 5,000 rpm at 4°C for 5 min. 50 μl of the supernatant was used for the detection of the expression of HBsAg S by enzyme-linked immunosorbent assay (ELISA). The ELISA kit was purchased from Shanghai Rongsheng Biotech Co., Ltd. China. Absorbance at 450 nm was measured and human serum-derived S Ag was used as a standard. Since preS2 gene and S gene compose HBsAg M gene and the two fragments are in the same open reading frame (ORF), detection of HBsAg S protein can represent the expression of HBsAg M protein in an ELISA assay.
To estimate the amount of CTB expressed in transgenic plants, proteins were extracted from 0.5 g of fresh leaf, stem, flower, and fruit tissue of the confirmed transgenic tomato plants or 0.5 g of fresh tissues of transgenic tobacco plants. Tissues were homogenized in liquid nitrogen and resuspended in 1 ml of extraction buffer (200 mM Tris–HCl (pH 8.0), 100 mM NaCl, 400 mM sucrose, 10 mM EDTA, 14 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, 0.05% Tween-20) and placed at 4°C overnight. The mixture was then centrifuged at 17,000 × g at 4°C for 15 min and the insoluble cell debris was removed. Standard rCTB (gift from Military Medical Scientific Institute, Beijing) was diluted into the concentration gradients of 1, 0.5, 0.25, 0.125, 0.063, and 0.032 μg/ml protein and the soluble protein samples were coated by coating buffer. 100 μl was used to coat each well of 96-well microtiter plate, and the plate was incubated overnight. The plate was then washed three times with PBS-T (PBS plus 0.05% Tween-20). The background was blocked with 1% (w/v) BSA solution in PBS at 37°C for 2 h, and the plate was washed three times with PBS-T. The plates were then incubated with monoclonal antibody of rabbit anti-CTB IgG that was diluted 1:1000 in 0.01 M PBS containing 0.5% BSA for 2 h at 37°C, followed by three washes with PBS-T buffer. Secondary labeling was done using horseradish peroxidase-conjugated goat anti-rabbit IgG (Takara) with 1:3000 dilution in 0.01 M PBS containing 0.5% BSA for 2 h at 37°C, followed by three washes with PBS-T buffer. The plates were developed by addition of 100 μl per well of tetramethylbenzidine (TMB) substrate (TakaRa) for 30 min at room temperature in dark. The plate was read at 492 nm using an ELISA reader (BioRad), and the amount of plant-expressed CTB was estimated based on the known amount of purified rCTB.
Results
Cloning and sequence analysis of E8 promoter
Tomato fruit-specific 1.1 kb- and 2.2 kb- E8 promoters were amplified by PCR from the genomic DNA isolated from L. esculentum cv. Jinfeng #1 (Fig. 2) and sequenced after subcloning into a TA-cloning vector pMD18-T. The whole promoter sequence has been deposited into GenBank with accession number DQ317599.

PCR amplification of tomato fruit-specific 1.1 kb- and 2.2 kb- E8 promoters Lane 1, Negative control of PCR; Lane 2, PCR products of tomato fruit-specific 2.2 kb E8 promoter fragment; lane 3, PCR products of tomato fruit-specific 1.1 kb E8 promoter fragment; and lane 4, DNA Marker DL2000
Nucleotide BLAST searches of the whole promoter sequence with published sequences were performed on NCBI (www. ncbi.nlm.nih.gov) (data not shown) revealed that the resultant tomato fruit-specific E8 promoter was highly conservative. The region −2159 to −1 which represents the 2.2 kb promoter is 99.9% and 99.4% identical to L. esculentum cv. VFNT Cherry (Deikman and Fischer 1988) and L. esculentum cv. Zhongshu No. 5 (GenBank submission number: AF515784. 2002), respectively. The region −1086 to −1 which represents the 1.1 kb promoter is 100% and 99.5% identical to VFNT Cherry and Zhongshu No. 5, respectively. The sequence differences of the promoter among Zhongshu No. 5, VFNT Cherry, and Jinfeng #1 were shown in Table 2.
Expression of HBsAg M protein in tobacco under the control of E8 promoter
The HBsAg M gene fragment was amplified by PCR from plasmid pADR-1 containing hepatitis B virus subtype adr. Plant expression vector pBI-1.1E8-S2S (Fig. 1), in which the 1.1 kb promoter-HBsAg M gene replaced the 35S promoter-gusA fragment of pBI121 at Hind III/Sac I sites and the HBsAg M gene was flanked downstream by the NOS-terminator, was constructed. The vector was then mobilized into A. tumefaciens LBA4404 by freeze-thaw method. Kanamycin-resistant Agrobacterium cells were confirmed by PCR and prepared for the transformation of N. tobacum cv. Xanthi.
Tobacco leaf-sections transfected by A. tumefaciens LBA4404 were placed on selection medium of MS agar with hormones (1 mg/l BA and 0.1 mg/l NAA) and antibiotics (100 mg/l kanamycin and 500 mg/l cefotaxime). Shoots began to develop on callus at the edge of the leaf-sections two weeks later. The shoots 1–2 cm in height were cut off from the callus and transferred to MS agar medium supplemented with 0.1 mg/l of NAA and 100 mg/l of kanamycin for rooting.
Twenty independent transgenic lines transformed with pBI-1.1E8-S2S were successfully obtained. The presence of the 1.1 kb E8 promoter-HBsAg M gene in the genomic DNA of the putative transgenic plants was initially examined by PCR. Sixteen out of twenty showed the expected 1.1 kb E8 promoter-HBsAg M gene band of ca. 1.9 kb in size. No band was amplified from wild-type plant.
To further confirm the integration of the HBsAg M in the transgenic tobacco genome, chromosomal DNA prepared from the 16 PCR-positive plants transformed with pBI-1.1E8-S2S were digested with Hind III/Sac I followed by Southern blot analysis. Plasmid pBI-1.1E8-S2S DNA and genomic DNA of untransformed plant were also digested by Hind III/Sac I and used as positive and negative control respectively. Southern hybridization showed the band with expected size in 14 out of 16 HBsAg M-transgenic lines, which confirmed the stable integration of the HBsAg M fragment into the tobacco genome. No hybridization band was detected in untransformed plant (Fig. 3).

Result of Southern hybridization of transgenic tobacco plants transformed with pBI-1.1E8-S2S containing E8-HBsAg M fragment. Lanes 1–7, genomic DNA of transgenic tobacco plants e1, e3, e5, e6, e14, e17, and e18 digested with Hind III + Sac I; Lane 8, genomic DNA of untransformed tobacco plant digested with Hind III + Sac I; and lane 9, plasmid pBI-1.1E8-S2S digested with Hind III + Sac I
ELISA assay was performed on crude protein extracts prepared from leaves, flowers, and seeds of 14 transgenic tobacco lines whose DNA samples were positive in Southern blot to examine for the expression of HBsAg S protein. Cutoff values were calculated for leaves, flowers, and seeds of untransformed plant respectively, where cutoff = OD450 (negative average) × 2.1. A sample was positive when the OD450 was higher than the cutoff value and was negative when less than the cutoff value. Results showed that no sample was positive for HBsAg S protein (Fig. 4), which indicated that there was no HBsAg S protein expression in tobacco even in tobacco seeds when the gene was controlled by the tomato fruit-specific E8 promoter. This suggests that the tomato fruit-specific E8 promoter has no biological function in tobacco.

HBsAg-ELISA results of transgenic tobaccos transformed with pBI-1.1E8-S2S and untransgenic plant (OD value). HBsAg M protein extracted from leaf, flower, and seed of untransformed plant and transgenic tobacco plant lines e6, e14, e17 and e18
Expression of HBsAg M protein in tobacco under the control of CaMV 35S promoter
A. tumefaciens LBA4404 carrying vector pBIS2S (Fig. 1), in which the HBsAg M gene replaced the gusA gene of pBI121 and was controlled by the cauliflower mosaic virus (CaMV) 35S promoter, was used to transform N. tobacum cv. Xanthi cells. Shoots began to develop on callus at the edge of the leaf-sections two weeks after transfection. 25 lines of transgenic seedlings with well-developed root system under selection of 100 mg/l of kanamycin were successfully obtained and transferred to soil.
Presence of the target gene in the genomic DNA of the 25 putative transgenic lines was initially identified by PCR analysis. As expected, A fragment of about 850-bp in size was amplified from 19 lines. No band was amplified from wild-type plant (data not shown). To further confirm the integration of HBsAg M gene into the tobacco genome, chromosomal DNA prepared from the PCR-positive plants was double digested with BamH I/Sac I followed by Southern blot analysis. Plasmid (pBIS2S) DNA and genomic DNA from non-transgenic plant were used as positive and negative control, respectively. Southern hybridization showed the band with expected size in all 19 transgenic lines (Fig. 5), which confirmed the stable integration of the HBsAg M gene into the tobacco genome.

Result of Southern hybridization of transgenic tobacco plants transformed with pBIS2S containing 35S-HBsAg M fragment.. Lanes 1–7, genomic DNA of transgenic tobacco plants s1, s2, s3, s11, s16, s17 and s19/BamH I + Sac I; lane 8, genomic DNA of non-transgenic tobacco plant/BamH I + Sac I; and lane 9, plasmid pBIS2S/BamH I + Sac I
Crude protein extracts prepared from leaves, flowers, and seeds of 10 random transgenic tobacco plants whose DNA samples were positive in Southern blots were used to examine the expression of HBsAg protein in transgenic tobacco plants by ELISA assay. A cutoff value was calculated according to the negative control (non-transgenic plant), where cutoff = OD450 (negative average) × 2.1, or cutoff = 0.05 when OD450 (negative average) was less than 0.05. A sample was regarded as positive when the OD450 was higher than the cutoff value. Results showed that 9 out of the 10 samples were positive for HBsAg S protein while the other one (line s15) was negative (Fig. 6).

Expression of HBsAg in transgenic tobacco lines via ELISA assay (OD value). s1, s2, …, and s19 represent independent transgenic lines. Cutoff value equals to multiplying the average OD450 value of nontransgenic plants by 2.1
Expression of CTB protein in tobacco under the control of E8 promoter
The plasmid pRTL-CTB (gift from Military Medical Scientific Institute, Beijing) was used to amplify the ctb gene. And plant expression vector pJES1 (Fig. 1), in which the 1.1 kb promoter-ctb gene replaced the 35S promoter-gusA fragment of pBI121 at Hind III/Sac I sites and the ctb gene was flanked downstream by the NOS-terminator, was constructed. Fifteen independent transgenic lines transformed with pJES1 were successfully obtained. The presence of the 1.1 kb E8 promoter-ctb gene fragment in the genomic DNA of the putative transgenic plants was initially checked by PCR. Twelve out of fifteen plants showed the expected E8 promoter-ctb gene band of ca. 1.5 kb in size. No band was amplified from wild-type plant.
To further confirm the integration of the ctb gene in the transgenic tobacco genome, chromosomal DNAs prepared from 12 Southern positive plants were digested with Hind III/Sac I followed by Southern blot analysis. Plasmid pJES1 DNA and genomic DNA of untransformed plant were digested by Hind III/Sac I and used as positive and negative control respectively. Southern hybridization showed the band with expected size in 9 out of 12 ctb-transgenic lines (data not shown). No hybridization band was detected in untransformed plant.
ELISA assay was performed on crude protein extracts prepared from fruit, leaf, stem and flower tissues of the 9 transgenic tobacco plants to examine the expression of CTB protein. No CTB expression in any transgenic plant was observed.
Expression of CTB protein in tomato under the control of E8 promoter
A. tumefaciens strain LBA4404 with plant expression vector pJES1, harbouring the ctb gene under the control of the 1.1 kb E8 promoter, was used to transform L. esculentum cv. Suifeng. Twenty-six kanamycin-resistant shootlets were obtained from the cotyledon callus and rooted after a series of treatment on elongating medium, rooting medium, and MS basic medium (kanamycin was added in all of above media to select the transformants). The rooted plantlets 5–10 cm in height were transferred to a greenhouse for fruiting.
14 independent transgenic tomato plants out of the 26 kanamycin-resistant plants were confirmed to have the exogenous ctb gene by using PCR (data not shown) and Southern blot analysis (Fig. 7).

Identification of ctb gene in transgenic tomato by Southern blotting (DNA was digested by Hind III and Sac I). Lane 1: plasmid pECT1; Lane 2: untransformed plant; Lane 3: negative transformed plant identified by PCR; Lanes 4–5: plant lines 9th and 16th
ELISA analysis of the protein extracted from fruit, leaf, stem and flower tissues of the 14 transgenic tomato plants for CTB expression demonstrated that only the ripe tomato fruits of 2 plant lines 9th and 16th had much higher OD value than the untransformed plant control, and no CTB expression was detected in leaf, stem, and unripe fruit even in lines 9th and 16th (Fig. 8). The level of CTB protein varied among the different developmental period of fruits of the same plant and presented an ascending tendency with the fruit ripening days. The highest level reached 0.455 and 0.385 μg/g fruit fresh weight in 31–40 d fruit tissues of transgenic tomato lines 9th and 16th respectively.

CTB-ELISA results of transgenic tomato plant lines 9th and 16th and untransgenic plant (OD value). X axis: samples of stem, leaf, flower, 0–10 d fruit, 11–20 d fruit, 21–30 d fruit, or 31–40 d fruit of tomato plant
Discussions
In recent years, plant has emerged as a convenient, safe and economical alternative to the mainstream expression system for pharmaceutical protein production including antibodies, vaccines, industrial enzymes, biopolymers and so on (Schillberg et al. 2005). Two species of plant, tobacco and tomato, hold their own advantages on this research area. Tobacco, with high biomass yields, robust transformation technology and strong biosafety profile, is at the forefront as an expression model for plant-based commercial protein production. Tomato is widely used for plant vaccines due to the feature of its fruit being palatable, nutritionally attractive, and could be eaten fresh.
The activity and specificity of a promoter, the main transcriptional regulator, can affect the level of transgene expression in plant. The E8 promoter has the fruit-specific expression feature, so it would be propitious to the growth of transgenic plant, harvest of the fruits, and the use of transgenic plant-made vaccine.
E8 promoter exists specially in tomato plants, and shows strong conservation in different tomato varieties. There were at least two components in the promoter contributing to its transcriptional regulation (Deikman and Fischer 1988; Deikman et al. 1992). The first is a downstream region of −1088 to the transcriptional start site of the promoter, which is sufficient for ripening-specific transcription in the absence of ethylene synthesis, and an element in the region from −1088 to −863 is required for high levels of expression during fruit ripening. The upstream region of −2181 to −1088 is known to contain ethylene-responsive transcriptional element (Deikman et al. 1992; Deikman et al. 1998). The 1.1 kb promoter used in this paper to control antigen expression in tobacco and tomato is 100% and 99.5% identical to its counterpart in L. esculentum cv. VFNT Cherry (Deikman and Fischer 1988) and cv. Zhongshu No. 5 (GenBank submission number: AF515784, 2002) respectively and was enough for gene expression. This implies that the E8 promoter can be widely used for gene expression in different varieties of transgenic tomato.
In order to explore the gene expression driven by E8 promoter in other plants beside tomato, we fused the 1.1 kb promoter to vaccine antigen HBsAg M gene to transform N. tabacum, whereas the CaMV 35S promoter was used for comparison. ELISA assay showed that the product of HBsAg M gene under the control of 1.1 kb promoter could not be detected in transgenic tobacco tissues such as leaves, flowers, and seeds. In contrast, the expression of HBsAg M gene driven by CaMV 35S promoter could be detected in transgenic tobacco. This result suggests that E8 promoter might act in a species-specific fashion.
To assess the utility of the tomato fruit-specific E8 gene’s promoter for driving vaccine antigen expression in tomato, we also fused the 1.1 kb promoter to cholera toxin B (ctb) gene to transform N. tabacum and L. esculentum respectively. ELISA assay for CTB proved that the 1.1 kb E8 promoter was able to direct the expression of exotic gene in ripe fruits of transgenic tomato, but absent in leaf, flower, and unripe fruit of tomato, which were consistent with that reported by Deikman and Fischer (1988), Good et al. (1994), Yakoby et al. (2006), and Mehta et al. (2002). However, no CTB protein was detected in transgenic tobacco tissues such as leaves, flowers, and seeds under the control of the 1.1 kb E8 promoter.
The DNA-binding activity of the E4/E8 binding protein (E4/E8BP) increased during fruit ripening of tomato, which suggested that this protein could play a role in regulation of gene expression (Deikman et al. 1998; Coupe and Deikman 1997). E4/E8BP is closely related to a DNA binding protein from tobacco, 3AF1 that interacts with the promoter of the pea rbcS-3A gene. A repeated domain within the 3AF1 amino acid sequence is conserved in E4/E8BP-1 (Coupe and Deikman 1997). Our result showed that no gene expression driven by the E8 promoter was observed in transgenic tobacco, whether this means certain protein(s) such as E4/E8-binding protein interacting specifically with certain element in the 1.1 kb promoter was specifically involved in tomato, but is absent in tobacco or other plants? Although tobacco DNA binding protein 3AF1 is homologous to tomato E4/E8 DNA binding protein E4/E8BP-1, and both are with similar affinity to recognition sequence (Coupe and Deikman 1997).
E8 gene expression is temporarily regulated by ethylene during fruit ripening. At the onset of fruit ripening, E8 mRNA showed an increase over the level of ethylene (Deikman and Fischer 1988). In our study, the expression of ctb gene in transgenic tomato fruits in the same plant presented an ascending tendency with the fruit ripening days, does this suggest that the 1.1 kb fragment of the E8 promoter is also responsive to the level of ethylene? Further researches are needed.
Abbreviations
- BA:
-
6-Benzylaminopurine
- CaMV 35S:
-
Cauliflower mosaic virus 35S
- CTB:
-
Cholera toxin B
- CTAB:
-
Cetyltrimethylammonium bromide
- EDTA:
-
Ethylenediamine tetraacetic acid
- ELISA:
-
Enzyme-linked immunosorbent assay
- HBsAg M:
-
Hepatitis B virus middle Antigen
- IAA:
-
Indolacetic acid
- MS :
-
Murashige and Skoog medium
- NAA:
-
α-Naphthaleneacetic acid
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- rCTB:
-
Recombinant cholera toxin B
- TMB:
-
Tetramethylbenzidine
References
Coupe SA, Deikman J (1997) Characterization of a DNA-binding protein that interacts with 5′ flanking regions of two fruit-ripening genes. Plant J 11:1207–1218
Deikman J, Fischer RL (1988) Interaction of a DNA binding factor with the 5′-flanking region of an ethylene-responsive fruit ripening gene from tomato. EMBO J 7:3315–3320
Deikman J, Kline R, Fischer R (1992) Organization of Ripening and Ethylene Regulatory Regions in a Fruit-Specific Promoter from Tomato (Lycopersicon esculentum). Plant Physiol 100: 2013–2017
Deikman J, Xu R, Kneissl ML, Ciardi JA, Kim KN, Pelah D (1998) Separation of cis elements responsive to ethylene, fruit development, and ripening in the 5′ flanking region of the ripening-related E8 gene. Plant Mol Biol 37:1001–1011
Doyle JJ, Doyle JL (1990) A rapid total DNA preparation procedure for fresh plant tissue. Focus 12:13–15
Gan RB, Chu MJ, Shen LP, Qian SW, Li ZP (1987) The complete nucleotide sequence of the cloned DNA of hepatitis B virus subtype adr in pADR-1. Sci Sin [B] 30:507–521
Good X, Kellogg JA, Wagoner W, Langhoff D, Matsumura W, Bestwick RK (1994) Reduced ethylene synthesis by transgenic tomatoes expressing S-adenosylmethionine hydrolase. Plant Mol Biol 26:781–790
Jani D, Meena LS, Rizwan-ul-Haq QM, Singh Y, Sharma AK, Tyagi AK (2002) Expression of cholera toxin B subunit in transgenic tomato plants. Transgenic Res 11:447–454
Krasnyanski SF, Sandhu J, Domier LL, Buetow DE, Korban SS (2001) Effect of an enhanced CaMV 35S promoter and a fruit-specific promoter on uidA gene expression in transgenic tomato plants. In Vitro Cell Dev Biol-Plant 37:427–433
Larrick JW, Thomas DW (2001) Producing proteins in transgenic plants and animals. Curr Opin Biotechnol 12:411–418
Mehta RA, Cassol T, Li N, Ali N, Handa AK, Mattoo AK (2002) Engineered polyamine accumulation in tomato enhances phytonutrient content, juice quality, and vine life. Nat Biotechnol 20:613–618
Sambrook J, Fritsch EF, Maniatis T (1992) Molecular cloning: a laboratory manual, (2nd edn). Cold Spring Harbor Press, New York
Sandhu JS, Krasnyanski SF, Domier LL, Korban SS, Osadjan MD, Buetow DE (2000) Oral immunization of mice with transgenic tomato fruit expressing respiratory syncytial virus-F protein induces a systemic immune response. Transgenic Res 9:127–135
Schillberg S, Twyman RM, Fischer R (2005) Opportunities for recombinant antigen and antibody expression in transgenic plants – technology assessment. Vaccine 23:1764–1769
Smigocki AC, Owens LD (1988) Cytokinin gene fused with a strong promoter enhances shoot organogenesis and zeatin levels in transformed plant cells. Proc Natl Acad Sci USA 85:5131–5135
Thanavala Y, Yang Y-F, Lyons P, Mason HS, Arntzen C (1995) Immunogenicity of transgenic plant-derived hepatitis B surface antigen. Proc Natl Acad Sci USA 92:3358–3361
Yakoby N, Garvey A, Raskin I (2006) Tobacco ribosomal DNA spacer element elevates Bowman–Birk inhibitor expression in tomato plants. Plant Cell Rep 25:573–581
Acknowledgements
This work was supported by The Research Foundation of Science and Technology Plan Project in Guangdong Province of China (Grant #2006B19901004) and The Natural Science Foundation of Guangdong Province of China (Grant #001212). Thanks are due to Dr. R. Sathishkumar at Department of Biotechnology, Bharathiar University, India and Dr. Qiuyun Liu at Biotechnology Research Center, Sun Yat-Sen University, China for providing valuable suggestions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhu-Mei He and Xiao-Ling Jiang contributed equally to this work.
Rights and permissions
About this article
Cite this article
He, ZM., Jiang, XL., Qi, Y. et al. Assessment of the utility of the tomato fruit-specific E8 promoter for driving vaccine antigen expression. Genetica 133, 207–214 (2008). https://doi.org/10.1007/s10709-007-9201-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-007-9201-2