
Abstract
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is an autosomal dominant condition in which susceptible individuals are at risk for the development of cutaneous leiomyomas, early onset multiple uterine leiomyomas and an aggressive form of type 2 papillary renal cell cancer. HLRCC is caused by germline mutations in the fumarate hydratase (FH) gene which inactivate the enzyme and alters the function of the tricarboxylic acid (Krebs) cycle. Issues surrounding surveillance and treatment for HLRCC-associated renal cell cancer were considered as part of a recent international symposium on HLRCC. The management protocol proposed in this article is based on a literature review and a consensus meeting. The lifetime renal cancer risk for FH mutation carriers is estimated to be 15 %. In view of the potential for early onset of RCC in HLRCC, periodic renal imaging and, when available, predictive testing for a FH mutation is recommended from 8 to 10 years of age. However, the small risk of renal cell cancer in the 10–20 years age range and the potential drawbacks of screening should be carefully discussed on an individual basis. Surveillance preferably consists of annual abdominal MRI. Treatment of renal tumours should be prompt and generally consist of wide-margin surgical excision and consideration of retroperitoneal lymph node dissection. The choice for systemic treatment in metastatic disease should, if possible, be part of a clinical trial. Screening procedures in HLRCC families should preferably be evaluated in large cohorts of families.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
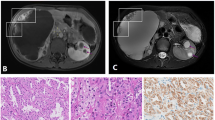
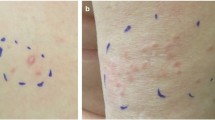
Hereditary leiomyomatosis and renal cell cancer (HLRCC) (OMIM #150800) is an autosomal dominant condition clinically characterized by three main features: (a) multiple cutaneous piloleiomyomas, which present as firm reddish skin papules or nodules, often in a segmental fashion, (b) multiple early-onset uterine leiomyomas which in many cases lead to myomectomies or hysterectomy and (c) an early-onset form of type 2 papillary renal cell cancer which has a propensity to metastasize early. The risk of renal cell cancer in HLRCC is uncertain due to the considerable variation of renal cell cancer prevalence among published cohorts of families [1–5]. The main clinical characteristics of HLRCC are illustrated in Fig. 1.

The following clinical criteria were proposed for a likely diagnosis of HLRCC: (1) histologically confirmed multiple cutaneous piloleiomyomas or (2) at least two of the following manifestations: surgical treatment for symptomatic uterine leiomyomas before age 40, type 2 papillary renal cell carcinoma before age 40 or a first-degree family member who meets one of these criteria [6]. Collecting duct renal carcinoma before age 40 has been suggested as an additional histological subtype indicative for the syndrome [7].
The gene alteration in HLRCC was identified in 2002 by Tomlinson et al. This research group identified underlying heterozygous germline mutations in the fumarate hydratase (FH) gene, which encodes the tricarboxylic acid (TCA, Krebs) cycle enzyme which catalyses the conversion of fumarate to malate [8]. In a subsequent study among 35 North American HLRCC families 31 (89 %) had germline FH mutations [9].
Pathogenic germline FH mutations have now been detected in 76–100 % of families with suggestive clinical features [7]. In families with characteristic features but without a demonstrated germline FH mutation the diagnosis HLRCC can be supported by immunohistochemical studies of tumors. In tumors with fumarate hydratase defects accumulated fumarate will lead to succination of proteins which can be revealed by an immunohistochemical assay [10].
Wider application of FH mutation analysis will likely reveal a more variable clinical picture of HLRCC than that observed thus far in the “classic” pedigrees. Remarkably, a recent study showed FH germline mutations in patients with paragangliomas [11].
The uncertain renal cell cancer risk in HLRCC, the documented childhood onset and the aggressive nature of many type 2 papillary renal cell cancers in HLRCC have raised questions concerning surveillance and treatment. We have considered these issues as part of the Fifth Symposium on Birt–Hogg–Dubé syndrome and Second Symposium on HLRCC, held in Paris, France, on June 28 and 29, 2013. The management protocol proposed in this article is based on a literature review and a consensus meeting. Recently, an international collaboration has been established for evaluation of renal cell cancer in HLRCC. The outcome of this evaluation may lead to higher levels of evidence for clinical recommendations in the future.
Clinical characteristics of renal cancer in HLRCC
Based on the distinct clinical, histological and cytological features of renal cell cancer in HLRCC [4, 5] this tumour type has recently been listed as a separate entity in the classification of renal neoplasia as “HLRCC-associated RCC” [12]. The histological picture is usually described as type 2 papillary renal cell cancer. It should be noted, however, that pathological features are variable and include a spectrum of architectural patterns. Importantly, other syndromic features in affected patients may be absent or inconspicuous. Therefore, the further development of the immunological assay described by Bardella et al. may yield an additional diagnostic tool in patients with suspected HLRCC [4, 5, 10, 13]. In Table 1 renal cancer prevalence figures are given for a series of studies. Renal cancer has been observed in about 20 % of families, but the prevalence figures vary greatly, probably largely due to variable ascertainment of kindreds. Figure 2 shows the distribution of age at diagnosis for 103 individuals with HLRCC-associated renal cell cancer for which age at diagnosis was reported. For this group the mean age at diagnosis was 41 years with a range from 11 to 90 years. Apparently, 7 % of cases have been diagnosed before the age of 20 years.

Distribution of ages at diagnosis of renal cell cancer in HLRCC in 103 patients reported in literature. References with number of patients between brackets: [1] (1), [13] (9), [17] (1), [2, 3] (1), [18] (12), [4] (37), [19] (1), [20] (2), [21] (1), [22] (1), [23] (3), [24] (1), [25] (1), [6] (1), [16] (25), [26] (1), [27] (1), [28] (1), [29] (1) [30] (2)
Among the larger series of cases [4, 16, 18] 3/74 (4 %) of patients had RCC before age 20 and this might indicate publication bias of very early onset cases.
The lifetime renal cancer risk in HLRCC is around 15 % based on expert opinion. The type of FH mutation does not seem to be an essential factor in renal cancer risk. In addition, there is no evidence that renal cancer risk is especially high in families in which renal cancer has occurred previously [8, 9, 31, 32].
In their study of 40 renal tumours resected from 38 patients belonging to HLRCC families with proven FH germline mutations Merino et al. [4] found the renal tumours to be predominantly unilateral and solitary. Treatment, follow-up and outcome for 19 patients with HLRCC-associated renal cancer was reported by Grubb et al. [5]. HLRCC patients whose tumours were detected and managed surgically when they were small have been observed to have a favourable prognosis with no evidence of disease after partial or radical nephrectomy for localized disease. However, the authors found the rate of distant metastasis in HLRCC to be higher than observed for other hereditary renal cancer syndromes such as Birt–Hogg–Dubé syndrome. Among the 27 patients with HLRCC-associated kidney cancer described by Gardie et al. [16] 20 (74 %) died due to metastatic disease. Since these patients were diagnosed after clinical symptoms had occurred the poor outcome in this group may reflect both delayed diagnosis and the aggressive nature of this renal cancer subtype.
Genetic (DNA) testing and renal cancer surveillance
In general, the pros and cons of surveillance in individuals with an increased cancer risk have to be weighed carefully, since surveillance not only has potential advantages (early detection and treatment of cancer) but also potential disadvantages. The efficacy of renal cancer surveillance in HLRCC has not yet been formally analysed and the rarity of HLRCC means that such an analysis would need to be performed either by a multicentre international collaboration or at centers with a large experience of this disorder.
DNA testing for late-onset hereditary tumour syndromes and subsequent surveillance of mutation carriers is generally performed from the age of 16–18 years onward, based on the assumption, that from this age, the individual can weigh the pros and cons of DNA testing without parental involvement. In contrast, DNA testing of children is advised for those conditions in which the identification of the familial mutation should lead to preventive measures starting in childhood.
In HLRCC the estimated cumulative renal cancer risk is about 15 % and based on the age distribution of RCC in this condition (Fig. 2), the risk for FH mutation carriers to develop renal cancer before age 20 years is an estimated 1–2 %.
The varying recommendations for surveillance as given in literature are summarized in Table 2. Based on the occurrence of renal cancer as early as 10 years old (Linehan, personal communication) childhood FH mutation testing and surveillance have been recommended for HLRCC from the age of 8 years onward (www.hlrccinfo.org) or even as early as 5 years of age [23]. Other groups apparently have concluded that for the young age group the risk is small and that DNA testing and surveillance could be postponed until the age of 18–20 years, thereby avoiding the burden of surveillance for young children.
We here propose that predictive FH mutation testing (when technically possible) should be offered prior to renal cancer surveillance in order to avoid unnecessary investigations. However, refusal of genetic testing should not prevent access to a surveillance programme. DNA testing should be considered from the age of 8–10 years onward but decided on an individual basis. While individuals who test negative for the familial pathogenic FH mutation do not need surveillance, mutation carriers should be offered a yearly MRI with 1–3 mm slices through the kidneys in order to detect very small tumours. If surveillance is started at a childhood age (after a positive FH mutation result) a baseline MRI taking 1–3 mm slices through the kidneys should be performed with subsequently annual MRI. Renal ultrasound is not recommended due to the low sensitivity of ultrasound to detect small lesions (unless ultrasound is the only available imaging modality).
Based on the present state of knowledge, decisions about predictive testing and surveillance before age 18 years should be made on an individual basis and the results of all procedures should be recorded to allow further audit and evaluation. We recommend strongly that the results of HLRCC screening investigations be collected prospectively and made available to appropriate research and audit projects.
Treatment of renal cancer
In the following two paragraphs, surgical curative treatment of localised renal cancer and palliative treatment of metastatic disease in HLRCC will be considered. For a general discussion of cytoreductive nephrectomy in metastatic renal cancer and the current status of adjuvant and neoadjuvant systemic therapy the reader is referred to a recent review [33].
Surgical curative treatment of renal cancer in HLRCC
About 25 years ago the standard surgical treatment for localised renal cancer was radical nephrectomy. At present, however, nephon sparing therapy is generally recommended for small renal tumours [34]. Nephron sparing therapy is especially important for patients with hereditary renal cancer which is associated with a predisposition to bilateral and multifocal tumours. Synchronous or metachronous renal cancers may call for multiple episodes of treatment and preservation of renal function is therefore an essential consideration in these patients. Thus in von Hippel–Lindau syndrome, hereditary papillary renal carcinoma and Birt–Hogg–Dubé syndrome, surgical intervention is recommended when the diameter of the largest tumour exceeds 3 cm. This “3 cm rule” is applied due to the relatively slow growth of the renal tumours in these syndromes [35–40]. However, HLRCC provides an exception to this rule since in this condition metastasis can occur even in cases of small primary tumours, which are often unilateral and solitary. HLRCC-associated kidney cancer represents a distinctive tumour type with a different molecular pathogenesis and a more aggressive biological behaviour than most other types of kidney cancer. Consequently, an active surveillance approach is not recommended for renal tumours in HLRCC patients.
In HLRCC, for the delineation of a tumour found at renal MRI, subsequent abdominal CT scanning may be recommended. Management of a suspected renal cancer most often consists of open partial nephrectomy. Prompt surgical extirpation with wide surgical margins and consideration of retroperitoneal lymphadenectomy are advised [5]. Radical surgery may be recommended if there is doubt that a partial nephrectomy would be curative. Neither radiofrequency ablation (RFA) nor cryotherapy for renal cancer is advised for patients affected with HLRCC.
Systemic treatment of metastatic renal cancer in HLRCC
Insight into the molecular pathogenesis of renal cancer has led to the development of new forms of “targeted” therapy aimed at intervention in the molecular pathways involved. Since clear cell renal cancer (ccRCC) is the most common histological subtype of renal cell cancer and since much of the molecular pathogenesis of this tumour type has been clarified, most forms of targeted therapy have been studied in cohorts of patients with sporadic ccRCC. At the molecular level, the most well-studied genetic pathways involved in the pathogenesis of ccRCC include the VHL tumour suppressor/hypoxia inducible factor (HIF) pathway [including HIF targets, such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF) and epidermal growth factor (EGF)] and the mTOR pathway. For common clear cell renal cancer there is renewed interest in the role of immunotherapy (notably the only potentially curative form of therapy for patients with advanced clear cell carcinoma) and a main role for approaches targeting the VHL/HIF pathway. In the US, the FDA has currently approved seven agents targeting the VHL/HIF and mTOR pathways, i.e. bevacizumab, pazopanib, axitinib, sunitinib, sorafenib, temsirolimus and everolimus [41, 42].
Recently, aberrant metabolism in renal cell cancer has been identified as an essential factor in kidney tumour formation and newer forms of targeted therapy are aimed at deranged metabolic pathways, in particular abnormalities of the TCA cycle and glycolysis [43, 44].
In HLRCC the main histological tumour type is papillary type 2 and a primary molecular alteration is a disruption of the TCA cycle. Germline mutations in the FH gene resulting in fumarate hydratase deficiency lead to a decrease in oxidative phosphorylation due to alteration of the function of the TCA cycle [45] and impairment of the oxidative function of the electron transport chain [46]. Accumulated fumarate functions as an oncometabolite. This effect is due to multiple mechanisms, including HIF stabilisation with subsequent effects on HIF targets and disrupted function of multiple proteins due to succination [47, 48]. In addition, metabolic abnormalities associated with FH inactivation are associated with epigenetic abnormalities [49, 50]. The pathways involved and the interaction between these pathways in HLRCC are manifold and complex and reviewed in several recent articles [51, 52].
At present, no standard therapy has been developed for patients with metastatic type 2 papillary RCC associated with germline FH mutations. Currently, a phase 2 study of bevacizumab and erlotinib targeting VEGF and EGFR, respectively, is open to subjects with advanced HLRCC-associated renal cancer or sporadic papillary RCC (www.clinicaltrials.gov). The rationale for this form of treatment is the presumed role of reduced fumarate hydratase activity in HIF stabilisation and activation of HIF targets.
Yamasaki et al. [26] described the case of a 24-year-old woman with HLRCC and metastatic papillary type 2 renal cancer, initially treated with a mTOR inhibitor. When progression of disease occurred after 5 months, 2-deoxy-d-glucose (2DG), an inhibitor of glycolysis, was administered based on the notion that inhibition of glycolysis might be especially effective for tumour cells with a TCA cycle defect since these tumours are dependent on glycolysis for ATP production. Unfortunately, however, this therapy had no clinical effect and further studies are needed to determine whether disruption of glycolysis in HLRCC-associated cancers will be feasible and effective. Various other forms of targeted therapy in HLRCC are currently being developed as summarized by Linehan and Rouault [52].
Summary and conclusion
The estimated lifetime renal cancer risk for FH mutation carriers is probably about 15 %. The exact cumulative risk figure should be determined in future studies. Currently, we cannot identify FH mutation carriers with a lower versus a higher renal cell cancer risk. As outlined above, no clear genotye–phenotype correlations have been found and there is no evidence for a lower RRC risk in kindreds with a negative family history for RCC. In view of the documented early onset of RCC in HLRCC, periodic renal imaging and, when available, predictive testing for a FH mutation should be offered starting from 8 to 10 years of age. However, the small risk of renal cell cancer in the 10–20 years age range and the potential drawbacks of screening should be carefully discussed on an individual basis in order to reach the optimum decision for each case. Imaging preferably consists of yearly renal MRI. Treatment of local tumours should be prompt since the 3 cm rule (see above) is not applicable for HLRCC. Treatment generally consists of wide-margin surgical excision and consideration of retroperitoneal lymph node dissection. The choice for systemic treatment in metastatic disease should preferably be part of a clinical trial. Current clinical trials including treatment of renal cell cancer in HLRCC are summarized at www.clinicaltrials.gov.
HLRCC exemplifies the challenges posed by rare diseases in which there is wide phenotypic variation ranging from minor skin involvement to fatal metastatic RCC. Clinicians have to balance the likely benefits and drawbacks of screening procedures with a paucity of evidence from prospective studies and an absence of biomarkers to indicate which FH mutation carriers are at especially high risk of RCC. Hence screening procedures in HLRCC families should preferably be evaluated in large cohorts of families through international collaboration.
References
Reed WB, Walker R, Horowitz R (1973) Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol 53:409–416
Alam NA, Rowan AJ, Wortham NC et al (2003) Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12:1241–1252
Alam NA, Barclay E, Rowan AJ et al (2005) Clinical features of multiple cutaneous and uterine leiomyomatosis. An underdiagnosed tumor syndrome. Arch Dermatol 141:199–206
Merino MJ, Torres-Cabala C, Pinto P et al (2007) The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31:1578–1585
Grubb RL III, Franks ME, Toro J et al (2007) Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177:2074–2080
Smit DL, Mensenkamp AR, Badeloe S et al (2011) Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 79:49–59
Lehtonen HJ (2011) Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 10:397–411
Tomlinson IPM, Alam NA, Rowan AJ et al (2002) The Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410
Toro JR, Nickerson ML, Wei M-H et al (2003) Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73:95–106
Bardella C, El-Bahrawy M, Frizzell N et al (2011) Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 225:4–11
Castro-Vega LJ, Buffet A, De Cubas AA et al (2013) Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 23:2440–2446
Srigley JR, Delahunt B, Eble JN et al (2013) The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol 37:1469–1489
Chen Y-B, Brannon AR, Toubaji A et al (2014) Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer. Recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol 38:627–637
Chuang GS, Martinez-Mir A, Engler DE et al (2005) Multiple cutaneous and uterine leiomyomata resulting from missense mutations in the fumarate hydratase gene. Clin Exp Dermatol 31:118–121
Wei M-H, Toure O, Glenn GM et al (2006) Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43:18–27
Gardie B, Remenieras A, Kattygnarath D et al (2011) Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet 48:226–234
Chan I, Wong T, Martinez-Mir A et al (2005) Familial multiple cutaneous and uterine leiomyomas associated with papillary renal cell cancer. Clin Exp Dermatol 30:75–78
Lehtonen HJ, Kiuru M, Ylisaukko-oja SK et al (2006) Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43:523–526
Lehtonen HJ, Blanco I, Piulats JM et al (2007) Conventional renal cancer in a patient with fumarate hydratase mutation. Hum Pathol 38:793–796
Al Refae M, Wong N, Patenaude F et al (2007) Hereditary leiomyomatosis and renal cell cancer: an unusual and aggressive form of hereditary renal carcinoma. Nat Clin Pract Oncol 4:256–261
Ghaninejad H, Moeineddin F, Rajaee A et al (2008) Hereditary leiomyomatosis and renal cell carcinoma syndrome: a case report. Dermatol Online J 14:16
Ahvenainen T, Lehtonen HJ, Lehtonen R et al (2008) Mutation screening of fumarate hydratase by multiplex ligation-dependent probe amplification: detection of exonic deletion in a patient with leiomyomatosis and renal cell cancer. Cancer Genet Cytogenet 183:83–88
Alrashdi I, Levine S, Paterson J et al (2010) Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Fam Cancer 9:239–243
Rongioletti F, Fausti V, Ferrando B et al (2010) A novel missense mutation in fumarate hydratase in an Italian patient with a diffuse variant of cutaneous leiomyomatosis (Reed’s syndrome). Dermatology 221:378–380
Onder M, Glenn G, Adisen E et al (2010) Cutaneous papules, uterine fibroids, and renal cell cancer: one family’s tale. Lancet 375:170
Yamasaki T, Tran TAT, Oz OK et al (2011) Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol 8:165–171
Raymond VM, Herron CM, Giordano TJ et al (2012) Familial renal cancer as an indicator of hereditary leiomyomatosis and renal cell cancer syndrome. Fam Cancer 11:115–121
Van Spaendonck-Zwarts KY, Badeloe S, Oosting SF et al (2012) Hereditary leiomyomatosis and renal cell cancer presenting as metastatic kidney cancer at 18 years of age: implications for surveillance. Fam Cancer 11:123–129
Behnes CL, Schlegel C, Shoukier M et al (2013) Hereditary papillary renal cell carcinoma primarily diagnosed in a cervical lymph node: a case report of a 30-year-old woman with multiple metastases. BMC Urol 13:3
Kuwada M, Chihara Y, Lou Y et al (2014) Novel missense mutation in the FH gene in familial renal cancer patients lacking cutaneous leiomyomas. BMC Res Notes 7:203
Bayley J-P, Launonen V, Tomlinson IPM (2008) The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9:20
Vahteristo P, Koski TA, Näätsaari L et al (2010) No evidence for a genetic modifier for renal cell cancer risk in HLRCC syndrome. Fam Cancer 9:245–251
Kennedy PA (2012) Wood CG Integration of surgery and systemic therapy for renal cell carcinoma. Urol Clin N Am 39:211–231
MacLennan S, Imamura M, Lapitan MC et al (2012) Systematic review of oncological outcomes following surgical management of localised renal cancer. Eur Urol 61:972–993
Walther MM, Choyke PL, Glenn G et al (1999) Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 161:1475–1479
Herring JC, Enquist EG, Chernoff A et al (2001) Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: 10-year experience. J Urol 165:777–781
Duffey BG, Choyke PL, Glenn G et al (2004) The relationship between renal tumor size and metastases in patients with von Hippel–Lindau disease. J Urol 172:63–65
Grubb RL III, Choyke PL, Pinto PA et al (2005) Management of von Hippel–Lindau-associated kidney cancer. Nat Clin Pract Urol 2:248–255
Pavlovich CP, Grubb RL III, Hurley K et al (2005) Evaluation and management of renal tumors in the Birt–Hogg–Dubé syndrome. J Urol 173:1482–1486
Joly D, Méjean A, Corréas JM et al (2011) Progress in nephron sparing therapy for renal cell carcinoma and von Hippel–Lindau disease. J Urol 185:2056–2060
Hu B, Lara PN Jr, Evans CP (2012) Defining an individualized treatment strategy for metastatic renal cancer. Urol Clin N Am 39:233–249
Ljungberg B, Bensalah K, Bex A et al (2013) Guidelines on renal cell carcinoma. Eur Assoc Urol. www.uroweb.org
Linehan WM, Srinivasan R, Schmidt LS (2010) The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 7:277–285
Linehan WM (2012) Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res 22:2089–2100
Mullen AR, Wheaton WW, Jin ES (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481:385–388
Tong W-H, Sourbier C, Kovtunovych G et al (2011) The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 20:315–327
Isaacs JS, Jung YJ, Mole DR et al (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153
Yang M, Soga T, Pollard PJ et al (2012) The emerging role of fumarate as an oncometabolite. Front Oncol 2:85
Xiao M, Yang H, Xu W et al (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326–1338
Kaelin WG Jr, McKnight SL (2013) Influence of metabolism on epigenetics and disease. Cell 153:56–69
Singer EA, Gupta GN, Srinivasan R (2012) Targeted therapeutic strategies for the management of renal cell carcinoma. Curr Opin Oncol 24:284–290
Linehan WM, Rouault TA (2013) Molecular pathways: fumarate hydratase-deficient kidney cancer—targeting the Warburg effect in cancer. Clin Cancer Res 19:3345–3352
Stewart L, Glenn GM, Stratton P et al (2008) Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch Dermatol 144:1584–1592
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Intramural Research Program of the NIH, Frederick National Laboratory, Center for Cancer Research. This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, NIH, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. The research was also supported by the Wellcome Trust Centre for Human Genetics, Grant Reference 090532/Z/09/Z. The Centre Expert National Cancers Rares PREDIR (S. Richard) is supported by grants from the French National Cancer Institute (INCa) and the French Department of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Menko, F.H., Maher, E.R., Schmidt, L.S. et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Familial Cancer 13, 637–644 (2014). https://doi.org/10.1007/s10689-014-9735-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-014-9735-2