
Abstract
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a tumor predisposition syndrome caused by heterozygous germline mutations in the fumarate hydratase (FH) gene. Cutaneous and uterine leiomyomas are the most common clinical manifestations of HLRCC, whereas only approximately 20% of the families display renal cell cancer (RCC). The number of RCC cases in these families varies from one to five. Interestingly, families with multiple RCC cases are mainly found in Finland and the USA. Such aggregation of RCC in only some families and populations has led to the hypothesis that besides FH mutations also other inherited genetic and/or environmental factors may contribute to the malignant kidney tumor formation. To search for such a genetic modifier we have performed a genome-wide linkage analysis in two and an identical by descent analysis in four Finnish HLRCC families with several RCC patients. Additional Finnish and French families were used in fine-mapping and haplotype analyses. The only region compatible with linkage was the locus surrounding the FH gene itself in chromosome 1q43. The genes in the putative candidate region were screened, but no potentially pathogenic alterations were observed. Although these data do not rule out the existence of a genetic modifier, they emphasize the contribution of the FH genotype in HLRCC related RCC. Therefore, as all FH mutation carriers may have an increased risk for developing renal cancer, counseling and genetic testing should be offered for all HLRCC family members and clinical follow-up should be organized for the mutation carriers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary leiomyomatosis and renal cell cancer (HLRCC, MIM 605839) is an autosomal dominant tumor predisposition syndrome characterized by cutaneous and uterine leiomyomas and renal cell cancer (RCC) [1, 2]. The predisposing gene is fumarate hydratase (FH) which encodes fumarase (FH), an enzyme of mitochondrial tricarboxylic acid cycle (TCAC, citric acid cycle, Kreb’s cycle) [3]. HLRCC patients have a heterozygous germline FH mutation, and in most cases the tumors harbor a second hit in FH [3–5]. To date, FH mutations have been described in more than 130 HLRCC families.
The most characteristic clinical features in HLRCC are cutaneous and uterine leiomyomas. RCC patients have been observed in only about 20% of the HLRCC families [1, 3, 6–15]. The number of RCC cases in these families varies from one to as many as five. These renal carcinomas mainly display papillary architecture and belong to a rare histological papillary type II subtype [1, 3, 5, 16]. A unique feature for HLRCC-related renal tumors is the presence of a large nucleus with a prominent eosinophilic nucleolus surrounded by a clear perinucleolar halo [1, 5]. A few HLRCC patients with collecting duct or conventional clear cell renal carcinoma have also been reported [5, 7, 8, 14, 17, 18]. RCC patients with a FH mutation are often diagnosed at younger age than sporadic RCC patients, and the tumors are associated with an aggressive metastatic disease with poor prognosis [1, 5, 18, 19].
Clustering of the RCC cases in only a small subset of families is typical for HLRCC. The majority of families with multiple renal cancers have been reported in Finland and the USA [1, 3, 7–15]. On the contrary, for example in the UK only one individual with RCC has been reported [7]. No clear correlation between the malignant phenotype and the type or site of the FH mutation or FH enzyme activity level has been verified [14, 20]. These observations have led to the suggestion that an environmental and/or genetic factor(s) that modify the effect of an underlying FH mutation may be required for the malignant kidney tumor development [3, 9]. Identification of such a factor would enable identification of individuals at high risk of developing aggressive RCC. This would enable directing possible screening procedures only to individuals at substantially increased risk, with the aim of early diagnosis and better clinical management of the patients. In this study, we utilized genome-wide genetic linkage and identical by descent (IBD) analyses in Finnish HLRCC families with multiple RCC cases to identify such a factor. In addition, a new, previously unidentified French family with multiple RCC cases as well several isolated RCC cases from HLRCC families from Europe and the USA were included in fine-mapping and candidate gene screening.
Patients and methods
Appropriate research permissions were obtained from local ethics committees. An informed consent was received from patients taking part of the study and, in case of deceased individuals, permission from appropriate authorities was obtained.
HLRCC families in linkage analysis
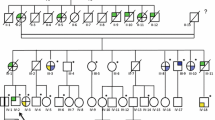
Altogether 22 members from two Finnish HLRCC families were included in the genome-wide linkage analysis (FAM-1 in Launonen et al. [1], FAM-5 in Lehtonen et al. [4], Fig. 1). These included seven RCC patients, four from FAM-1 and three from FAM-5. In FAM-1 there are five RCC cases, four of which have been diagnosed with papillary type II RCC at ages 33, 34, 39, and 42 years. All these four patients are females and included in the analysis. During this project, the fifth FH mutation positive RCC patient was diagnosed with collecting duct carcinoma at the age of 52 years. In FAM-5 there are four RCC patients, of whom three with the histology of papillary type II RCC were included in the analysis. Of these, the father has been diagnosed at the age of 71 years, and his two daughters at 36 and 49 years of age. The fourth RCC patient in the family is an aunt of the father, and has been diagnosed with clear cell renal cancer at the age of 90 years. Due to the atypical HLRCC histology, lack of the second FH hit in the tumor, and late age at disease onset the patient was not included in the linkage analyses.

Pedigrees of the studied HLRCC families. Only patients with RCC are marked as affected. Ages at RCC diagnosis are presented below individuals’ symbols. Recently diagnosed RCC patients are marked with a thick downward arrow (gray) and index persons are indicated by a thin upward arrow (black). The index person of the FAM-1 does not have RCC and is not shown in the figure. The pedigrees have been slightly modified for confidentiality
In addition to families in genome-wide linkage analysis, 18 additional samples were available for fine-mapping. These included a patient from FAM-1 recently diagnosed with collecting duct carcinoma, as well as two additional families with several RCC cases (Fig. 1). FAM-2 is a Finnish family with three siblings diagnosed with papillary type II RCC, a brother diagnosed at the age of 26 years and two sisters diagnosed at 32 and 51 years of age (FAM-2 in Launonen et al. [1]). FAM-F is a previously unreported French FH mutation positive (G397R) family with three RCC patients. The father had died of RCC (histology unknown), and his two sons had undergone surgery due to papillary type II RCC at the age of 59 years. One of the sons had died of metastasis 2 years after the operation.
RCC patients in mutation analyses
Genomic DNA samples from FH mutation positive RCC patients were included in the mutation analyses. At least one RCC case from each known Finnish HLRCC family (n = 5) was included in all candidate gene searches. In addition, as new families from other populations were identified (n = 6) during the course of the study, members from these families were also included in the analyses. These include one individual from Swedish [21], Spanish [17], UK and US families each, as well as members from two French families. Latter three families are novel HLRCC kindreds not previously reported in the literature. In the US family, the index patient had been diagnosed with a metastatic clear cell RCC at the age of 32 years, and had died of the disease 4 years later. The patient and his father had both had cutaneous leiomyomas, and the paternal grandfather had died of RCC of unknown histology at the age of 64 years. FH mutation R101X was found from the index patient. One of the French families is the previously mentioned FAM-F, and the other is an isolated RCC patient who had been diagnosed with a papillary type II tumor at the age of 33 years and deceased 1 year later. The FH mutation of this patient is IVS-2A>G.
Linkage analysis
DNA samples from 17 members from FAM-1 and five members from FAM-5 were genotyped by deCODE (Reykjavik, Iceland) using 2000 microsatellite markers. Allegro software [22] was used for the genome-wide linkage and haplotype analysis using data from the most informative individuals assuming dominant inheritance. RCC patients were defined as affected, FH mutation positive healthy relatives as healthy, and FH mutation negative relatives as unknown. HaploPainter (http://haplopainter.sourceforge.net/) [23] was used to draw haplotypes.
Fine-mapping with additional microsatellite repeats and SNPs was performed at the most promising loci using members from the two families in genome-wide linkage analyses, as well as 18 additional samples. The data were analyzed using SimWalk2, Allegro, and Merlin [24, 25].
SNP array and IBD analysis
In order to detect possible shared IBD haplotypes between RCC cases in different families we used data from high resolution SNP panels. DNA samples from five RCC cases from four Finnish HLRCC families were genotyped using Affymetrix (Santa Clara, CA, USA) Human Mapping 50 K Xba and/or 50 K HindIII SNP arrays. Additionally, SNP array data from altogether seven 1st degree relatives were used for haplotype phasing. Samples from all Finnish RCC cases and their 1st degree relatives from whom high quality DNA from a fresh tissue sample (solid tissue or blood) was available were used. Minimum criteria for IBD in this analysis (shared haplotype size >1 Mb, number of SNPs in a fragment >30) were set based on random sampling of data from our other SNP genotyping projects. IBD analysis and haplotyping was done using Merlin package tools.
Direct sequencing of the candidate genes
The coding regions and exon–intron boundaries of the candidate genes were sequenced using standard PCR protocols and ABI3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The primer sequences and specific PCR conditions are available from the authors upon request.
Results and discussion
A genome-wide linkage and haplotype analysis with an average microsatellite marker density of 2 cM resulted in positive logarithm of odds (LOD) score on 12 chromosomal regions in 10 chromosomes (Table 1). After fine-mapping and haplotype analyses chromosomal regions 1q23, 3, 4, 16 and 19 were excluded due to recombinations (FAM-1, FAM-5). Candidate genes were sequenced from the remaining regions according to plausible function as a putative cancer gene (n = 47). Members from different families were included in the mutation analyses to maximize the mutation detection by the screening method. However, no disease-associated genetic alterations were identified. Subsequently, a new RCC case was diagnosed in FAM-2 and samples from a new French HLRCC family became available for the analyses. With these additional samples all regions except 22q13 and the locus surrounding the FH gene in 1q43 were excluded. All the five genes in chromosome 22 region were screened with negative results. At this stage of the study, the fifth RCC case in FAM-1 was diagnosed. This individual was found not to share the chromosome 22 region. Thus, the only remaining region compatible with linkage was the FH locus itself.
To study the possibility of a FH-linked modifier, we further examined the chromosome 1q43 region. The putative candidate region was defined by two criteria: First, the modifier should reside within a region where two Finnish families with altogether eight RCC cases and the same FH mutation (FAM-1, FAM-2) share a common haplotype. In addition to FH, there are six genes in this region: muscarinic acetylcholine receptor M3 (CHRM3), formin 2 (FMN2), gremlin-2 precursor (GREM2), regulator of G protein signalling 7 (RGS7), kynurenine 3-monooxygenase (KMO), and opsin 3 (OPN3) (Fig. 2). Second, a strong modifier with a tumor suppressive function would likely reside outside an area where three UK HLRCC families carry large germline deletions [3, 7] as these families have no RCC patients. Only three genes fulfill both of these criteria: CHRM3, FMN2, and GREM2 (Fig. 2). All coding regions of these three genes were sequenced but no mutations were observed. There is a small possibility that the modifier is located for instance in a promoter region or in intronic sequence, or that it is a relatively common SNP that could be detected only by association studies with much larger sample sets.

Schematic presentation of the genes residing in the possible modifier locus very close to the FH gene. FH fumarate hydratase, CHRM3 muscarinic acetylcholine receptor M3, FMN2 formin 2, GREM2 gremlin-2 precursor, RGS7 regulator of G protein signalling 7, KMO kynurenine 3-monooxygenase, OPN3 opsin 3
To further explore potential modifier candidate loci, a SNP array analysis was performed. Samples from five Finnish RCC cases and their 1st degree relatives (n = 7) were used. With these SNP panels (100,000 or 50,000 SNPs per sample) we aimed to detect common IBD haplotypes shared by RCC cases. IBD could potentially carry a founder mutation or a common variation undetectable by conventional linkage analysis. In our preliminary IBD scan, two regions with potential extended shared IBD were found (1.4 and 2.8 Mb in chromosomal regions 2p16 and 11q14, respectively). However, detailed haplotype dissection revealed no reliable common haplotypes among the RCC patients in neither of these regions. Although no convincing IBD was detected, the existence of smaller shared fragments carrying a potential modifier allele(s) cannot be excluded as they may be out of the detection capacity of the analysis.
Despite these efforts, we found no evidence of a genetic modifier for RCC in the context of HLRCC syndrome. Even though our results do not exclude the possible existence of a genetic modifier, this suggests that FH inactivation alone may be the key event in malignant kidney tumor formation. Results from a recently created HLRCC mouse model support this conclusion [26]. Kidney-specific Fh1 mutants developed multiple abnormally large clonal renal cysts which are suggested to be premalignant lesions. In humans, kidney cysts have been observed in HLRCC patients more frequently and at younger age than in control individuals [4]. Moreover, inactivation of Fh1 in the mouse kidney has been shown to cause activation of the hypoxia pathway, which is suggested to be the mechanism behind HLRCC-related RCC tumorigenesis [26]. A diminished FH activity leads to pseudohypoxia via accumulation of FH substrate fumarate. Fumarate inhibits hypoxia-inducible factor (HIF) prolyl hydroxylases from hydroxylating HIFs [27]. The unhydroxylated HIFs, which are transcription factors upregulating the transcription of genes associated with e.g. angiogenesis and glycolysis [27], are stabilized in HLRCC renal tumors [27, 28].
The same HIF-related mechanism is also behind succinate dehydrogenase complex (SDH) -deficient tumorigenesis [27–29]. SDH is one of the TCA cycle enzyme and germline mutations in subunits SDHB/C/D have been shown to cause hereditary paragangliomatosis (TCA Cycle Gene Mutation Database (2004) http://chromium.liacs.nl/lovd_sdh/home.php [30]). Interestingly, mutations in the SDHB gene have also been reported to predispose to RCC both in the context of familial paraganglioma [31–33] as well as in patients with no personal or family history of pheochromocytoma or head and neck paraganglioma but with familial or bilateral RCC [34]. These observations support the notion that mutations in TCAC genes may themselves be sufficient for RCC formation.
In summary, we found no evidence of a genetic factor that would modify the RCC risk in the context of HLRCC syndrome. Although this does not rule out the existence of a genetic modifier, it suggests that all FH mutation carriers may have an increased risk for developing renal cancer. Typically these tumors are aggressive and associate with poor prognosis. Thus it is important to offer counseling and genetic testing for all HLRCC family members, and clinical follow-up should be organized for the mutation carriers to assure early detection of possible malignancies. In Finland, for example, contrast enhanced MRI with a 6 month interval is recommended for all FH mutation positive patients over 18 years. Encouraging example of this strategy was recently shown when the third patient in FAM-2 (Fig. 1) was diagnosed with an aggressive but early stage RCC during a regular screening, and the tumor was successfully surgically removed.
Abbreviations
- CHRM3 :
-
Muscarinic acetylcholine receptor M3
- cM:
-
Centimorgan
- FH :
-
Fumarate hydratase (gene)
- FH:
-
Fumarase (enzyme)
- FMN2 :
-
Formin 2
- GREM2 :
-
Gremlin-2 precursor
- HIF:
-
Hypoxia-inducible factor
- HLRCC:
-
Hereditary leiomyomatosis and renal cell cancer
- IBD:
-
Identical by descent
- LOD:
-
Logarithm of odds
- Mb:
-
Mega base pairs
- MRI:
-
Magnetic resonance imaging
- RCC:
-
Renal cell cancer
- SDH:
-
Succinate dehydrogenase complex
- SNP:
-
Single nucleotide polymorphism
- TCAC:
-
Tricarboxylic acid cycle
References
Launonen V, Vierimaa O, Kiuru M et al (2001) Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98(6):3387–3392
Alam NA, Bevan S, Churchman M et al (2001) Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3–q43. Am J Hum Genet 68(5):1264–1269
Tomlinson IP, Alam NA, Rowan AJ et al (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30(4):406–410
Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK et al (2006) Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43(6):523–526
Merino MJ, TorresCabala C, Pinto P et al (2007) The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31(10):1578–1585
Kiuru M, Launonen V, Hietala M et al (2001) Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol 159(3):825–829
Alam NA, Rowan AJ, Wortham NC et al (2003) Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12(11):1241–1252
Toro JR, Nickerson ML, Wei MH et al (2003) Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73(1):95–106
Alam NA, Olpin S, Rowan A et al (2005) Missense mutations in fumarate hydratase in multiple cutaneous and uterine leiomyomatosis and renal cell cancer. J Mol Diagn 7(4):437–443
Chan I, Wong T, Martinez-Mir A et al (2005) Familial multiple cutaneous and uterine leiomyomas associated with papillary renal cell cancer. Clin Exp Dermatol 30(1):75–78
Chuang GS, Martinez-Mir A, Geyer A et al (2005) Germline fumarate hydratase mutations and evidence for a founder mutation underlying multiple cutaneous and uterine leiomyomata. J Am Acad Dermatol 52(3 Pt 1):410–416
Chuang GS, Martinez-Mir A, Engler DE et al (2006) Multiple cutaneous and uterine leiomyomata resulting from missense mutations in the fumarate hydratase gene. Clin Exp Dermatol 31(1):118–121
Badeloe S, van Geel M, van Steensel MA et al (2006) Diffuse and segmental variants of cutaneous leiomyomatosis: novel mutations in the fumarate hydratase gene and review of the literature. Exp Dermatol 15(9):735–741
Wei MH, Toure O, Glenn GM et al (2006) Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43(1):18–27
Refae MA, Wong N, Patenaude F et al (2007) Hereditary leiomyomatosis and renal cell cancer: an unusual and aggressive form of hereditary renal carcinoma. Nat Clin Pract Oncol 4(4):256–261
Alam NA, Olpin S, Leigh IM et al (2005) Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol 153(1):11–17
Lehtonen HJ, Blanco I, Piulats JM et al (2007) Conventional renal cancer in a patient with fumarate hydratase mutation. Hum Pathol 38(5):793–796
Grubb RL 3rd, Franks ME, Toro J et al (2007) Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177(6):2074–2079
Alam NA, Barclay E, Rowan AJ et al (2005) Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141(2):199–206
Bayley JP, Launonen V, Tomlinson IP et al (2008) The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9:20
Ahvenainen T, Lehtonen HJ, Lehtonen R et al (2008) Mutation screening of fumarate hydratase by multiplex ligation-dependent probe amplification: detection of exonic deletion in a patient with leiomyomatosis and renal cell cancer. Cancer Genet Cytogenet 183(2):83–88
Gudbjartsson DF, Jonasson K, Frigge ML et al (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25(1):12–13
Thiele H, Nurnberg P (2005) HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 21(8):1730–1732
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58(6):1323–1337
Abecasis GR, Cherny SS, Cookson WO et al (2002) Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30(1):97–101
Pollard PJ, Spencer-Dene B, Shukla D et al (2007) Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell 11(4):311–319
Isaacs JS, Jung YJ, Mole DR et al (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8(2):143–153
Pollard PJ, Briere JJ, Alam NA et al (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14(15):2231–2239
Selak MA, Armour SM, MacKenzie ED et al (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7(1):77–85
Bayley JP, Devilee P, Taschner PE et al (2005) The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet 6:39
Vanharanta S, Buchta M, McWhinney SR et al (2004) Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 74(1):153–159
Neumann HP, Pawlu C, Peczkowska M et al (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292(8):943–951
Srirangalingam U, Walker L, Khoo B et al (2008) Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin Endocrinol (Oxf) 69(4):587–596
Ricketts C, Woodward ER, Killick P et al (2008) Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100(17):1260–1262
Acknowledgments
Sini Marttinen, Iina Vuoristo, Inga-Lill Svedberg, Mikko Aho, and Anniina Raitila are warmly thanked for excellent technical assistance. The study has been supported by grants from European Commission (LSHC-CT-2005-518200), Academy of Finland (Center of excellence in Translational Genome-Scale Biology and grants 212901, 214323, 213183, 214268), Finnish Cancer Society, the French National Cancer Institute (Kidney PNES, INCa) and the French League against Cancer (Comités du Cher et de l’Indre).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors Pia Vahteristo and Taru A. Koski have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Vahteristo, P., Koski, T.A., Näätsaari, L. et al. No evidence for a genetic modifier for renal cell cancer risk in HLRCC syndrome. Familial Cancer 9, 245–251 (2010). https://doi.org/10.1007/s10689-009-9312-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-009-9312-2