
Abstract
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC) is a hereditary condition which typically presents with cutaneous and uterine leiomyomata. Papillary type II renal cell carcinoma and other less common histologic subtypes of renal cancer have been reported in HLRCC. We describe the case of a 31-year-old man in which the pathology review of his renal carcinoma and a positive family history of renal carcinoma allowed for the identification of a pathogenic mutation in the FH gene (c.698G>A;p.R233H) confirming the diagnosis of HLRCC. Recognition of this syndrome allowed for appropriate surveillance as well as identification of at-risk family members. Pathology review is essential for accurate diagnosis of a hereditary cancer syndrome in the setting of familial renal cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary Leiomyomatosis and Renal Cell Carcinoma, HLRCC, (MIM: 150800) is an autosomal dominant condition which predisposes to the development of cutaneous and uterine leiomyomas and early-onset renal cell carcinoma [1]. The condition was described in its first reports as Multiple Cutaneous and Uterine Leiomyomata, MCUL (MIM: 150800) [2–4]. Despite the fact that the original patient described by Reed et al. [2] had a personal history of renal cancer diagnosed at age 20, it was only later that the association between leiomyomata and renal cancer was established [5]. HLRCC is caused by germline mutations in the fumarate hydratase (FH) gene [1].
Cutaneous leiomyomata are often the primary presenting clinical feature with over 80% of individuals with HLRCC developing these tumors [6, 7]. Uterine leiomyomata, while common in the general population, develop earlier in women with HLRCC and tend to be larger in quantity and size and can aid in making the diagnosis of this hereditary condition [8]. Cutaneous and uterine leiomyosarcomas have also been reported in FH mutation carriers. Cutaneous leiomyosarcomas were found in 1 out of 81 patients (1.2%) with a clinical or molecular diagnosis of HLRCC in one clinical series [7], and 1 in 46 patients (2.2%) with an FH mutation in a second series [8]. In a population-based series from Finland, mutations in FH were found in 1 out of 67 (1.5%) women with nonsydromic uterine leiomyosarcoma [9].
The renal cancers in HLRCC have been described as papillary type II carcinomas. These HLRCC-associated renal tumors have abundant cytoplasm, large nuclei with an eospinophilic nucleous surrounded by a clear halo [5, 10]. Lehtonen describes this as an “owl-eye like” nucleoli [11]. Additionally, these tumors are typically of a high Fuhrman grade 3/4 [5, 10, 11]. Other renal cancer types such as collecting duct carcinomas, sarcomatoid, oncocytic, tubulo-papillary, clear cell renal cancers as well as a case of Wilms’ tumor [6, 8, 10, 12, 13] have also been described in families with HLRCC. HLRCC-associated renal cancers tend to be unilateral and very aggressive in nature but bilateral cases have been reported [10, 14].
It is estimated that about 20-25% of HLRCC families report diagnoses of renal carcinoma [1, 7, 8, 11, 12, 14–17]. It is important to note that these estimates vary greatly, from 2.4% [16] in the United Kingdom to up to 62% [8] of US families seen at the NCI describing a family history of renal cancer [1, 7, 8, 11, 12, 14–17]. Ascertainment bias makes it difficult to accurately estimate the frequency from the literature, but 20–25% seems to be a reasonable summary of the prevalence. Penetrance has not yet been formally calculated in any published study.
Fumarate hydratase is an enzymatic member of the tri-carboxycylic acid cycle (Krebs’ cycle), and catalyzes the conversion of fumarate to malate. The gene also functions as a tumor suppressor gene, with the second hit being loss of the wild type allele in a manner that conforms to classical loss of heterozygosity [18]. While individuals heterozygous for a mutation in FH are at-risk for features of HLRCC, individuals who are homozygous or compound heterozygous for germline FH mutations develop a very different phenotype, Fumarate Hydratase Deficiency (FHD). FHD (MIM: 606812) is a metabolic disorder which is associated with encephalopathy, developmental delay and death within the first few years of life [19, 20]. Leiomyomatosis has not been reported in patients with FHD, however parents of children with FHD have incidentally been found to have leiomyomatosis [1]. No malignant phenotype has been described in FHD families to date.
Here we report a case of HLRCC in a young man in which the pathology of his renal carcinoma in conjunction with the pathology of his father’s renal carcinoma led to the diagnosis of this hereditary cancer syndrome as his physical exam and family history were negative for cutaneous or uterine leiomyomata.
Patient and methods
Permission for research was obtained from the Institutional Review Board at the University of Michigan, Ann Arbor, USA. Written informed consent was received from the patients or their next of kin in the case of deceased individuals.
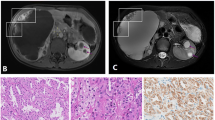
The patient is a 31-year old male who presented with left sided flank pain. A computed tomography scan of the chest, abdomen and pelvis was notable for a 6 cm × 4.7 cm left midpole renal mass with an adjacent hematoma. He underwent left nephrectomy demonstrating a 6.5 cm Fuhrman grade 3/4 papillary type II renal cell carcinoma (Fig. 1A, B). Surgical margins were free of tumor and the tumor was found to be invading into the perirenal fat, but not through gluteus fascia (T3a). No enlarged lymph nodes were identified. There was a large 11.5 cm subcapsular hemorrhage surrounding the mass and extending from the upper pole to the lower pole of the kidney. The mass grossly appeared necrotic, although there was no microscopic necrosis. The patient had postoperative imaging which demonstrated no evidence of residual disease within the abdomen and no evidence of pulmonary nodules, even though a 3 mm nodular density in the right lower lobe had been noted on the initial pre-operative radiology report. He was referred for a cancer genetics consultation due to his young age at cancer diagnosis.

Pathology of renal cancers arising in a family with HLRCC. A and B Pathology of proband (III:3). Type 2 papillary renal cell carcinoma with a papillary architecture. (A) and high-grade nuclear features (B). C and D Pathology of father (II:4). Poorly-differentiated renal cell carcinoma most consistent with a collecting duct carcinoma with predominant solid areas (C) and minority areas with tubular differentiation (D). H&E; original magnification ×200
During the genetics consultation, a physical exam of the proband was notable for multiple cherry angiomas, including two recently enlarging angiomas measuring 7–8 mm on the chest, and three atypical nevi. These were biopsied and found to be capillary hemangiomas and atypical (dysplastic) compound nevi and atypical (dysplastic) junctional nevi with slight and moderate atypia. There were no clinically suspicious cutaneous leiomyomas evident on physical exam and no evidence of leiomyomatosis in the biopsied lesions.
A detailed family history was obtained (Fig. 2) and was notable for poorly differentiated renal cell carcinoma, most consistent with collecting duct carcinoma diagnosed in the proband’s father at age 47, who died of metastatic disease at age 48 (Fig. 1C, D). This histopathology was confirmed by a second pathologist (TJG). After specific inquiry, the patient and his mother denied any history of cutaneous leiomyomas in the proband’s father. Maternal family history was unremarkable for cancer. The full maternal and paternal family histories were also negative for cutaneous or uterine leiomyoma after specific inquiry. There were no additional reported cases of renal carcinoma beyond the proband and his father.

Pedigree of Family 5194; Proband, denoted with arrow (III:4) presented at age 31 with Papillary type II RCC. Father (II:4) died at age 47 from a poorly differentiated RCC, likely a collecting duct cancer
A surgical specimen containing both tumor and normal tissue from the proband’s father was obtained. The DNA used for molecular analyses in this individual was obtained through microdissection of unstained re-cut slides of paraffin-embedded tumors using previously described methods [21]. Briefly, designated areas of normal and tumor tissue were determined by a board-certified pathologist (TJG) on a hematoxylin and eosin-stained slide. The designated areas on the corresponding unstained re-cut slides were removed with a clean razor blade and transferred to individual tubes. The DNA was then extracted from the scraped tissue by dissolving the samples in xylene to remove the paraffin followed by proteinase K digestion and ethanol precipitation.
Mutation detection within the proband’s father’s tumor was carried out by PCR and direct sequencing of DNA extracted from formalin fixed and paraffin embedded tumor tissue. PCR primers for the amplification of FH exon 5 were as follows: forward, 5′-TTT TCC CAC AGC AAT GCA C-3′; and reverse, 5′-TGC CCA AGA GTA AGT GGA ACA-3′. The reaction mix per sample included 10pmol each of the forward and the reverse primer along with a master mix made up of 2.5mM dNTPs (NEB), MgCl2, 10× PCR buffer, and Amplitaq Gold (Applied Biosystems). The PCR conditions begin with an initial denaturing step at 95°C for 10 min followed by 38 cycles of 94°C for 30 s, 60°C for 45 s, and 72°C for 45 s, and a final extension at 72°C for 10 min. To check the quality of the amplification, approximately 3μl of PCR product was run a 1.5% agarose gel by electrophoresis and then visualized by ethidium bromide. The PCR product was then purified using an Exo-SAP protocol which involves incubating the PCR product in 0.73μl of SAP buffer, 0.73 μl of SAP and 0.58 μl Exonuclease Restriction Enzyme. Dideoxy sequencing was performed by the sequencing core facilities at the University of Michigan (Ann Arbor, MI, U.S.A.). Sequences were compared to reference sequence NM_000143 using Mutation Surveyor version MS2.61 (SoftGenetics Inc), and each chromatogram was manually reviewed to confirm the mutation. Sequencing was performed in both directions to confirm sequence variations.
Results
Given the history of papillary type II renal cancer and family history of collecting duct carcinoma, genetic testing for mutations in the FH gene was ordered through a CLIA-certified clinical laboratory. Sequencing identified a missense mutation, p.R233H (c.698G>A) previously described as p.R190H (c.569G>A) [22]. This mutation is a well described, relatively frequent mutation in families with HLRCC [18]. Thus mutational analysis confirmed the suspected clinical diagnosis of HLRCC in the proband.
Analysis in tumor and normal tissue DNA of the proband’s father confirmed the presence of the p.R233H mutation (Fig. 3).

Chromatograms of Wild-Type and Mutant FH exon 5. Control sequence for (A) was derived from CEPH NA11873. B and C represent sequence from individual II:4, derived from normal adjacent kidney tissue and poorly differentiated renal carcinoma tissue, respectively. B and C Illustrate the single base substitution c.698G>A which results in substitution of Arginine by Histidine at codon 233 (p.R233H). Note that (B) shows presence of wild-type sequence (black), while (C) does not, consistent with loss of heterozygosity
Discussion
A diagnosis of Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC) in this family was suggested by the specific pathologic features of papillary type II renal cancer and collecting duct carcinoma and was confirmed by molecular genetic testing. About 20–25% of HLRCC families report a family history of renal cancer [1, 7, 8, 11, 12, 14–17]. The common presenting features of the condition, cutaneous and uterine leiomyomata, were absent in this family. Without proper pathology review of the renal tumors, a diagnosis of HLRCC would not have been possible.
Gardie et al. [12]. recently examined a series of 79 families identified either through a dermatology (N=56) or urology (N=23) clinic as having histories suggestive of HLRCC. Forty-four of 79 families had germline FH gene mutations (55.7%) including two of 5 families (40%) with familial isolated papillary type II renal cancer in the absence of other features of HLRCC and two of 18 families (11.1%) with sporadic papillary type II renal cancer in the absence of other feature of HLRCC. Fifteen of 44 families (34%) reported a family history of renal cancer and 27 of 151 (17.9%) known gene carriers had a personal history or renal cancer. Thirty-one unique FH mutations were identified in these 44 families and no genotype-phenotype correlation could be observed in regards to cutaneous or uterine leiomyomatosis, leiomyosarcoma or renal cancer risk. Genetic modifiers for renal cancer risk have not been identified [23].
HLRCC genotype-phenotype correlations have yet to be clearly established. Toro et al. [7] studied 31 families with a clinical diagnosis of HLRCC and a confirmatory mutation in FH. They found that 5 families reported histories of renal cell carcinoma, most often papillary type II. Missense mutations at the same codon (p.R233L; p.R233C) have been reported in association with renal cancer [22]. However, several mutations, including the p.R233H identified in this family, have been reported in HLRCC families with and without renal cancer [24] suggesting variable expressivity for specific mutations like p.R233H.
Toro, et al. [7] found that 11 of 35 (31.4%) unrelated families carried the p.R233H mutation and that it may represent a mutational hotspot. It is estimated that 18% of all reported FH mutations are at codon 233 [24]. The FH mutation database constructed by Bayley et al. [22] reports 19 independent families with this mutation described as the most common FH mutation.
When reviewing differential diagnoses for familial renal cancers, the tumor pathology provides vital clues to aid in the diagnosis (Table 1). The identification of the appropriate hereditary renal cancer predisposition syndrome has significant implications on surgical management and future surveillance as well as screening recommendations for at-risk relatives [25–27]. Renal cancers in individuals with von Hippel-Lindau (vHL), Hereditary Papillary Renal Carcinoma (HPRC) and Birt-Hogg-Dubé (BHD) are typically treated conservatively with cryoablation and nephron sparing surgery due to the likelihood of multifocal, bilateral disease and the relatively predictable clinical course of these cancers [28, 29]. The main objective in these particular inherited renal cancer predisposition syndromes is to preserve as much renal tissue as possible for long-term renal function. Recently, renal cancer has been found to be associated with Hereditary Paraganglioma Syndrome (HPGL), an inherited condition predisposing to head, neck and thoracic paraganglioma and adrenal and extra-adrenal pheochromoctyoma [30, 31]. The extent of surgical resections recommended for the management of renal cancer arising within the context of HPGL is not yet firmly established [32].
In contrast, individuals with HLRCC-associated renal carcinoma should be treated more aggressively, and these patients often undergo radical nephrectomy given the aggressive nature and highly metastatic potential of these cancers [33]. Individuals with HLRCC are less likely to develop bilateral tumors [10], although a single case with bilateral tumors has been reported [14]. The more typical, unilateral presentation favors aggressive resections to minimize the possibility of recurrence rather than less aggressive surgery that spares normal renal tissue in order to preserve long-term renal function.
The identification of HLRCC in this particular family is also representative of the utility of genetic testing and screening of at-risk relatives. The autosomal dominant inheritance pattern of HLRCC means that all first-degree relatives are at 50% risk of the condition. The average age of onset of renal cancer is 44 years in the North American HLRCC population and 43 years in the French HLRCC population [7, 12]. We recommend that at-risk individuals should be screened with annual abdominal/pelvic MRI, although some authors recommend either CT or MRI [24]. Once a lesion concerning for renal cancer is identified, intervals in screening should be decreased and follow-up evaluation with FDG-PET scans is recommended. When the concerning lesion reaches radiographic or clinical criteria that are not yet well defined, radical nephrectomy is recommended due to the aggressive nature of renal cancers seen in HLRCC. Although there are no clearly defined radiographic criteria, a mass that clearly appears to be a tumor, even when less than 1 cm in size, should be immediately removed, possibly with radical nephrectomy. Indeterminate lesions and complex cysts should be monitored with shorter intervals than annual surveillance. Given the recent reports of young onset renal cancer in an individual with HLRCC, predictive genetic testing and asymptomatic surveillance has been suggested as early as age 5 [34]. We advocate for predictive genetic testing and asymptomatic screening for HLRCC associated renal tumors beginning at age 18 with annual abdominal/pelvic MRI. As the patient ages, it is reasonable to consider incorporating abdominal/pelvic CT alternating with abdominal/pelvic MRI annually, although our preference is to minimize radiation and rely on MRI.
Management of cutaneous and uterine leiomyomata is symptomatic, however the risk of leiomyosarcoma needs to be considered. Screening and management guidelines are evolving and range from screening with annual transvaginal ultrasound and strong consideration of prophylactic hysterectomy once childbearing is complete to a more conservative approach with the consideration of hysterectomy as needed for management of uterine leiomyomas [11, 35].
In this family, the ability to make a clinical diagnosis of HLRCC was diminished by the relatively few at-risk female family members in a condition where one of the primary features is uterine leiomyomata. Here we demonstrate the importance of pathology review in an accurate risk assessment and differentiating this condition from the other hereditary cancer syndromes in the presence of familial renal cancer. A detailed review by an experienced pathologist is essential in making the diagnosis and aiding in decisions of surgical and medical management.
Abbreviations
- HLRCC:
-
Hereditary leiomyomatosis and renal cell carcinoma
- MCUL:
-
Multiple cutaneous and uterine leiomyomata
- vHL:
-
von Hippel-Lindau syndrome
- BHD:
-
Birt-Hogg-Dubé syndrome
- HPGL:
-
Hereditary paraganglioma syndrome
- HPRC:
-
Hereditary papillary renal carcinoma
References
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30(4):406–410. doi:10.1038/ng849ng849
Reed WB, Walker R, Horowitz R (1973) Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol 53(5):409–416
Rudner EJ, Schwartz OD, Grekin JN (1964) Multiple cutaneous leiomyoma in identical twins. Arch Dermatol 90:81–82
Fisher WC, Helwig EB (1963) Leiomyomas of the skin. Arch Dermatol 88:510–520
Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA (2001) Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98(6):3387–3392. doi:10.1073/pnas.051633798051633798
Smit DL, Mensenkamp AR, Badeloe S, Breuning MH, Simon ME, van Spaendonck KY, Aalfs CM, Post JG, Shanley S, Krapels IP, Hoefsloot LH, van Moorselaar RJ, Starink TM, Bayley JP, Frank J, van Steensel MA, Menko FH (2011) Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 79(1):49–59. doi:10.1111/j.1399-0004.2010.01486.x
Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, Stewart L, Duray P, Tourre O, Sharma N, Choyke P, Stratton P, Merino M, Walther MM, Linehan WM, Schmidt LS, Zbar B (2003) Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73(1):95–106. doi:AJHG035018
Wei MH, Toure O, Glenn GM, Pithukpakorn M, Neckers L, Stolle C, Choyke P, Grubb R, Middelton L, Turner ML, Walther MM, Merino MJ, Zbar B, Linehan WM, Toro JR (2006) Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43(1):18–27. doi:10.1136/jmg.2005.033506
Ylisaukko-oja SK, Kiuru M, Lehtonen HJ, Lehtonen R, Pukkala E, Arola J, Launonen V, Aaltonen LA (2006) Analysis of fumarate hydratase mutations in a population-based series of early onset uterine leiomyosarcoma patients. Int J Cancer 119(2):283–287. doi:10.1002/ijc.21798
Merino MJ, Torres-Cabala C, Pinto P, Linehan WM (2007) The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31(10):1578–1585. doi:10.1097/PAS.0b013e31804375b8
Lehtonen HJ (2011) Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 10(2):397–411. doi:10.1007/s10689-011-9428-z
Gardie B, Remenieras A, Kattygnarath D, Bombled J, Lefevre S, Perrier-Trudova V, Rustin P, Barrois M, Slama A, Avril MF, Bessis D, Caron O, Caux F, Collignon P, Coupier I, Cremin C, Dollfus H, Dugast C, Escudier B, Faivre L, Field M, Gilbert-Dussardier B, Janin N, Leport Y, Leroux D, Lipsker D, Malthieu F, McGilliwray B, Maugard C, Mejean A, Mortemousque I, Plessis G, Poppe B, Pruvost-Balland C, Rooker S, Roume J, Soufir N, Steinraths M, Tan MH, Theodore C, Thomas L, Vabres P, Van Glabeke E, Meric JB, Verkarre V, Lenoir G, Joulin V, Deveaux S, Cusin V, Feunteun J, Teh BT, Bressac-de Paillerets B, Richard S (2011) Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet 48(4):226–234. doi:10.1136/jmg.2010.085068
Badeloe S, van Spaendonck-Zwarts KY, van Steensel MA, van Marion AM, van Essen AJ, Jonkman MF, Steijlen PM, Poblete-Gutierrez P, van Geel M, Frank J (2009) Wilms tumour as a possible early manifestation of hereditary leiomyomatosis and renal cell cancer? Br J Dermatol 160(3):707–709. doi:10.1111/j.1365-2133.2008.09000.x
Lehtonen HJ, Blanco I, Piulats JM, Herva R, Launonen V, Aaltonen LA (2007) Conventional renal cancer in a patient with fumarate hydratase mutation. Hum Pathol 38(5):793–796. doi:10.1016/j.humpath.2006.10.011
Chuang GS, Martinez-Mir A, Engler DE, Gmyrek RF, Zlotogorski A, Christiano AM (2006) Multiple cutaneous and uterine leiomyomata resulting from missense mutations in the fumarate hydratase gene. Clin Exp Dermatol 31(1):118–121. doi:10.1111/j.1365-2230.2005.01977.x
Alam NA, Barclay E, Rowan AJ, Tyrer JP, Calonje E, Manek S, Kelsell D, Leigh I, Olpin S, Tomlinson IP (2005) Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141(2):199–206. doi:10.1001/archderm.141.2.199
Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, Salovaara R, Herva R, Koivisto PA, Vierimaa O, Aittomaki K, Pukkala E, Launonen V, Aaltonen LA (2006) Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43(6):523–526. doi:10.1136/jmg.2005.036400
Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP, Barclay E, Calonje E, Manek S, Adams SJ, Bowers PW, Burrows NP, Charles-Holmes R, Cook LJ, Daly BM, Ford GP, Fuller LC, Hadfield-Jones SE, Hardwick N, Highet AS, Keefe M, MacDonald-Hull SP, Potts ED, Crone M, Wilkinson S, Camacho-Martinez F, Jablonska S, Ratnavel R, MacDonald A, Mann RJ, Grice K, Guillet G, Lewis-Jones MS, McGrath H, Seukeran DC, Morrison PJ, Fleming S, Rahman S, Kelsell D, Leigh I, Olpin S, Tomlinson IP (2003) Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12(11):1241–1252
Zinn AB, Kerr DS, Hoppel CL (1986) Fumarase deficiency: a new cause of mitochondrial encephalomyopathy. N Engl J Med 315(8):469–475. doi:10.1056/NEJM198608213150801
Rustin P, Bourgeron T, Parfait B, Chretien D, Munnich A, Rotig A (1997) Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim Biophys Acta 1361(2):185–197. doi:S0925-4439(97)00035-5
Greenson JK, Bonner JD, Ben Yzhak O, Cohen HI, Miselevich I, Resnick MB, Trougouboff P, Tomsho LD, Kim E, Low M, Almog R, Rennert G, Gruber SB (2003) Phenotype of microsatellite unstable colorectal carcinomas: well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol 27(5):563–570
Bayley JP, Launonen V, Tomlinson IP (2008) The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9:20. doi:10.1186/1471-2350-9-20
Vahteristo P, Koski TA, Naatsaari L, Kiuru M, Karhu A, Herva R, Sallinen SL, Vierimaa O, Bjorck E, Richard S, Gardie B, Bessis D, Van Glabeke E, Blanco I, Houlston R, Senter L, Hietala M, Aittomaki K, Aaltonen LA, Launonen V, Lehtonen R (2010) No evidence for a genetic modifier for renal cell cancer risk in HLRCC syndrome. Fam Cancer 9(2):245–251. doi:10.1007/s10689-009-9312-2
Kiuru M, Launonen V (2004) Hereditary leiomyomatosis and renal cell cancer (HLRCC). Curr Mol Med 4(8):869–875
Linehan WM (2009) Genetic basis of bilateral renal cancer: implications for evaluation and management. J Clin Oncol 27(23):3731–3733. doi:10.1200/JCO.2009.23.0045
Singer EA, Bratslavsky G, Middelton L, Srinivasan R, Linehan WM (2011) Impact of genetics on the diagnosis and treatment of renal cancer. Curr Urol Rep 12(1):47–55. doi:10.1007/s11934-010-0156-y
Maher ER (2011) Genetics of familial renal cancers. Nephron Exp Nephrol 118(1):e21–e26. doi:10.1159/000320892
Dharmawardana PG, Giubellino A, Bottaro DP (2004) Hereditary papillary renal carcinoma type I. Curr Mol Med 4(8):855–868
Pavlovich CP, Grubb RL 3rd, Hurley K, Glenn GM, Toro J, Schmidt LS, Torres-Cabala C, Merino MJ, Zbar B, Choyke P, Walther MM, Linehan WM (2005) Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol 173(5):1482–1486. doi:10.1097/01.ju.0000154629.45832.30
Henderson A, Douglas F, Perros P, Morgan C, Maher ER (2009) SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam Cancer 8(3):257–260. doi:10.1007/s10689-009-9234-z
Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C (2004) Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 74(1):153–159. doi:10.1086/381054
Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, Cole TR, Armstrong R, Kumar VK, Morrison PJ, Atkinson AB, Douglas F, Ball SG, Cook J, Srirangalingam U, Killick P, Kirby G, Aylwin S, Woodward ER, Evans DG, Hodgson SV, Murday V, Chew SL, Connell JM, Blundell TL, Macdonald F, Maher ER (2010) Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31(1):41–51. doi:10.1002/humu.21136
Refae MA, Wong N, Patenaude F, Begin LR, Foulkes WD (2007) Hereditary leiomyomatosis and renal cell cancer: an unusual and aggressive form of hereditary renal carcinoma. Nat Clin Pract Oncol 4(4):256–261. doi:10.1038/ncponc0773
Alrashdi I, Levine S, Paterson J, Saxena R, Patel SR, Depani S, Hargrave DR, Pritchard-Jones K, Hodgson SV (2010) Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Fam Cancer 9(2):239–243. doi:10.1007/s10689-009-9306-0
Kiuru M, Launonen V, Hietala M, Aittomaki K, Vierimaa O, Salovaara R, Arola J, Pukkala E, Sistonen P, Herva R, Aaltonen LA (2001) Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol 159(3):825–829
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raymond, V.M., Herron, C.M., Giordano, T.J. et al. Familial renal cancer as an indicator of hereditary leiomyomatosis and renal cell cancer syndrome. Familial Cancer 11, 115–121 (2012). https://doi.org/10.1007/s10689-011-9485-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-011-9485-3