
Abstract
Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome (HLRCC) is a rare disease and since the first report, it has been found in just over 200 families approximately, around the world (Smit et al. in Clin Genet 79:49–59, 2009). Patients in Colombia or in Latin America have not been described, as far as we know. HLRCC is inherited in an autosomal dominant manner, and it is caused by heterozygous germline mutations in the FH gene, which encodes the fumarate hydratase enzyme. It is characterized mainly by the appearance of cutaneous and uterine leiomyomas, and an early-onset, aggressive form of type 2- papillary renal cell carcinoma (Smit et al. in Clin Genet 79:49–59, 2009; Schmidt and Linehan in Int J Nephrol Renovasc Dis 7:253–260, 2014]. We report a Colombian family with HLRCC syndrome, with a novel mutation in FH gene (c.1349_1352delATGA) in which cutaneous leiomyomas have not been found, but other clinical manifestations such as type 2- papillary renal cell carcinoma, uterine leiomyomas and rare tumors were present. This investigation constitutes the first report of HLRCC syndrome in Colombia, and probably in Latin America.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome (HLRCC OMIM 150800) is a rare disease, with an autosomal dominant pattern of inherence. Several families with the condition have been reported, especially in North America [3], the United Kingdom, the Netherlands, and Finland [1]. Descriptions of affected subjects have also been reported in India, Japan [1], and Spain [4].
HLRCC syndrome is caused by heterozygous germline mutations in the FH gene (1q42.3-43), which is responsible for encoding the fumarate hydratase protein. This enzyme catalyzes the conversion of fumarate in malate during the tricarboxylic acid cycle [5, 6]. It is believed that the inactivation of this enzyme involves an accumulation of the fumarate that acts as an oncometabolite, both in the mitochondria and in the cytoplasm [2, 7]. High concentrations of fumarate produce an over-activation of the pathways regulated by hypoxia inducible factor 1 alpha (HIF1-a) [6] and the factor erythroid 2-related factor 2 (NRF2) [8]. The over-expression of these genes could be responsible for the malignant transformation of the cells of HLRCC patients with renal cell cancer [6, 8]. Additionally, it has been found that the levels of the energy cell sensor, AMP-activated protein kinase (AMPK) are decreased [6].
Regarding the clinical manifestations, patients can present: cutaneous leiomyomas, which appear in 75 % of the cases at an average age of 25 years old. These skin erythematous lesions -papules or nodules- may increase in size and number with age and may be painful or cause paresthesia. They are usually located in the trunk or the limbs [2].
On the other hand, more than 70 % of affected women develop uterine leiomyomas, which are associated with severe pelvic pain, irregular menses, and menorrhagia. Usually these symptoms lead to an early hysterectomy [3]. Finally, 10–16 % of the patients will exhibit type 2-papillary renal cell carcinoma that has an early onset before the age of 40 and is characterized for being a very aggressive form of cancer with metastasis even during early stages. The tumors are often unilateral and have poor prognosis [1, 9]. Histologically, they are identified by a large eosinophilic nucleus with a clear perinuclear halo [1].
In 2011, Smit et al. [1] proposed a set of diagnostic criteria for HLRCC. The main criterion is the presence of multiple cutaneous leiomyomas, with histologically confirmation; minor criteria include: early onset type 2- papillary renal cell carcinoma, history of surgical treatment for multiple symptomatic uterine leiomyomas diagnosed before the age of 40 and a first degree family member who meets one of the mentioned criteria [1]. In general terms, diagnosis is likely in the presence of the main criterion, and the syndrome may be suspected when a patient meets at least two minor criteria [1]. Diagnosis is considered definite when germline mutation of FH is proved [2].
Nevertheless, the fact that patients with HLRCC present cutaneous lesions, which are mostly asymptomatic, uterine leiomyomas that are frequent in general female population and renal tumors that appear in less than 20 % of the cases, greatly complicates the diagnosis of this syndrome [1]. Diagnosis in Latin American countries is even more difficult given the scarcity of data related to renal cancer; because of that, knowledge about molecular aspects, diagnostic approaches, treatment, and genetic counseling must be improved in this region.
The aim of this article was to perform a clinical and genetic characterization of a Colombian family with findings compatible with HLRCC.
Case description
Ethical approval was obtained from the Ethics Committee of the Faculty of Medicine - Universidad Nacional de Colombia (Resolution Number 002-013-15). Recommendations of the Declaration of Helsinki with later amendments and the Belmont Report were followed. Informed consent was obtained from all the individual participants included in this study. In the case of children, informed assent was also obtained.
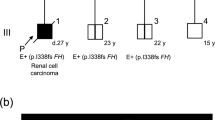
A 36-year-old man with history of partial right nephrectomy at the age of 35 arrived at the genetic consult He denied any particular symptom and only referred that the mass in the right kidney was observed in a routine abdominal computed tomography (CT) scan. The later biopsy confirmed a type 2- papillary renal cell carcinoma. His mother died due to the same type of cancer at the age of 58, as well as other family members (Fig. 1). The physical exam did not reveal anything unusual, and cutaneous leiomyomas were not found.

Family pedigree. II-2 and II-13 were cousins; information of the family of II-13 is not available. Subject IV-2 is the proband, affected with type 2- papillary renal cell carcinoma. Patient III-9 was positive for FH mutation and he had Sertoli cells tumor and basal cell carcinoma. Asterisks indicate which individuals were tested for FH mutation. Hereditary leiomyomatosis and renal cell cancer syndrome in a Colombian Family. This figure was created using Paint X lite program and Power Point program
HLRCC syndrome was considered as the first diagnostic possibility in the proband; analysis of germline mutations in FH was requested. This was performed in a certified international laboratory. The result was the evidence of 4 base pair deletion located in exon 9, named according to the Human Genome Variation Society guidelines (FH gene number of access: NM_000143.3) as c.1349_1352delATGA, which at protein level is referred to as p.Asn450SerfsX3 (counting from the first ATG of non-processed protein). This mutation had not been reported before and according with Variant Effect Predictor (VEP) platform by Ensembl, it is a frameshift mutation that produces a change of asparagine to serine in the position 450, creating a stop codon at the position 3 of the new reading frame. Such mutation was considered pathogenic because is predicted to have a high impact on normal protein function through protein truncation.
With the confirmation of a mutation in FH gene, family members at risk were incorporated in the investigation. DNA was extracted from peripheral blood, using a QIAamp DNA Blood Mini Kit by Qiagen following the manufacturer’s recommendations. Specific primers were designed to flank the mutation region using Primer3 and Primer-BLAST programs and we did the amplification by Polymerase Chain Reaction (PCR). Primers and PCR conditions are available under request.
Purification and sequencing of PCR products were performed by Macrogen Inc. (Seoul, Korea). The analysis of bidirectional sequences was performed using Mutation Surveyor Demo software (version 5.0; SoftGenetics, State College, PA) and CodonCode Aligner Demo software (version 602; CodonCode Corporation, Dedham, MA).
This study was conducted with 20 participants, including the proband (Fig. 1). 55 % were female and 45 % were male; the average age of men was 36.4 years old, and in the case of women, 41.1 years old. The ages ranged between 12 and 67 years old for females, and 13 and 65 years old for male subjects.
The pedigree of the family is shown in Fig. 1, in which according to the patients, 8 individuals had died due to metastatic renal cancer at early ages (II-2, II-5, II-9, II-10, II-11, II-12, III-1, III-11), although cutaneous leiomyomas were not documented in any of those cases. Regarding, female patients (II-2, II-5, II-11, III-1) it is not known for sure if they had uterine leiomyomas.
The heterozygous mutation found in the proband was also found in 5 more family members: subjects III-3, III-9, III-10, IV-1 and IV-18 (Fig. 2). All of them initiated a surveillance program (10, HLRCC Family Alliance; National Center Institute; http://www.hlrccinfo.org).

Partial Sequence chromatogram of the FH gene (exon 9). An example of the mutation found in all the patients. a A Subject with the Wild Type sequence (WT). b Patient IV-18 with the mutation c.1349_1352delATGA. The red line shows the position of the deletion, and the blue line shows the frameshift. This Figure was created using the images of the sequence results from CodonCode Aligner Demo software and also we use Power point program to add lines and words. (Color figure online)
Table 1 summarizes the main characteristics of patients with the mutation.
Cutaneous leiomyomas were not evidenced during the physical exam of any of the patients who were positive for FH mutation.
Given the existence of case reports of HLRCC syndrome with type 2- papillary renal cell carcinoma in patients under 8 years old [10, 11] we decided to perform the genetic test in the children following the guidelines of the American Academy of Pediatrics [12], but none of them had a positive result.
Discussion
This study characterized and reported the first family with HLRCC syndrome in Colombia, in which also a novel mutation in the FH gene was observed.
This is a relevant finding since, as we have stated before, HLRCC syndrome it is a rare disease, with no incidence data [13]. Specifically in Colombia, statistics about renal cancer are limited and there is no an established difference between sporadic and hereditary cases. This report may enrich clinical knowledge about the pathology in this part of the world.
The studied family has a novel mutation and contrary to the majority of descriptions in literature, it does not present cutaneous leiomyomas characteristic of this disease [1–3]. Nevertheless, it presents one of the least common but more devastating findings: the type 2- papillary renal cell carcinoma [1–3] that can be present in 20 % of the families [10]. However, as mentioned by Menko et al. [10], the type of mutation in FH seems not to be a determining factor in renal cancer risk.
The mutation reported in this article is a 4 base pair deletion that caused a frameshift, and compromise exon 9 of the FH gene. It creates a premature stop codon and the result is a truncated protein, so we assume, this would significantly affect normal protein function.
At first it seems logical to assume that the more deleterious a FH mutation is the greater the functional impact in the enzyme and the accumulation of fumarate, which could somehow, be reflected in a more aggressive tumor phenotype. Nonetheless, it is not possible to make a definite statement about this and, as discussed by other authors [3, 5, 14, 15], there is no clear association between FH mutations and cancer severity. Thus, more studies that clarify more aspects of the genotype-phenotype relation of this pathology are needed.
In the present case, we can only mention that the mutation was observed in a family with two predominant manifestations of HLRCC: renal cell cancer and uterine leiomyomas. However, these were not the only significant findings.
Two of the patients with the mutation had ultrasound and abdominal CT scans with small bilateral renal cysts, evidenced in one individual at the age of 28 and in the other at the age of 65 (Table 1).
The fact that female patient IV-3 (Fig. 1) who tested negative for FH mutation has also bilateral renal cysts, only illustrates, first, how common these findings can be in adults (prevalence of 10 % increasing with age [16]) and second, the reason why the presence of renal cysts is not considered a sensitive finding in the clinical diagnosis of HLRCC. However, in our opinion, it should be screened in mutation carriers, in part due to the presumption that these lesions could be the first stage of carcinogenesis in renal tissue [9].
It is also important to be cautious with uterine leiomyomas, which usually appear before the age of 40, as it was observed in women who tested positive, in comparison to the female patient who did not present the mutation and whose symptoms started after the age of 50.
We wanted to establish if there had been other types of cancer in the family besides renal cell carcinoma. In the pedigree (Fig. 1), two women of the first generation died due to cancer, but their location or type are unknown. The only individual with reliable data was III-9, who has the FH mutation and presented pathology reports that showed a basal cell carcinoma and a Sertoli cells tumor, both in remission.
During the literature review, we found that in this disease, other low frequency tumors besides uterine leiomyosarcoma have been described, such as: gastrointestinal stromal tumors (GIST) [1, 17], adrenal gland [1, 18], breast [19], bladder [19], and testicle tumors, specifically Leydig cells [20], not Sertoli. Regarding basal cell carcinoma, Lehtonen reports two patients in 2006 [19] with this type of cancer, who were also positive for FH mutation. The fact that a patient of this family exhibits two rare types of cancer in the context of HLRCC syndrome is an unusual and interesting finding.
Beyond rare or non-specific clinical signs, we have to mention that the family who participated in the study is part of the group of patients who daily have to suffer because of the little knowledge of their condition among medical staff.
Conclusions
This investigation reports a Colombian family with HLRCC Syndrome, a disease that could be underdiagnosed [1, 13] specially in this country and others in Latino America. Raise awareness and improve the knowledge in physicians and specialists of the existence of this syndrome, should be the first step to provide a better attention to the patients and their families, who suffer greatly by the absence of a clear diagnosis.
Abbreviations
- HLRCC:
-
Hereditary leiomyomatosis and renal cell cancer
- NRF2 :
-
Factor erythroid 2-related factor 2
- AMPK :
-
AMP-activated protein kinase
- PCR:
-
Polymerase chain reaction
- PHD:
-
Prolyl hydroxylases
- HIF-1a :
-
Hypoxia inducible factor 1 alpha
- 2SC:
-
2 Succinyl-cysteine
- CT:
-
Computed tomography
- WT:
-
Wild Type sequence
- GLUT1 :
-
Glucose transporter 1
- VEGF :
-
Vascular endothelial growth factor
- KEAP 1 :
-
Kelchlike ECH associated protein 1
- ROS:
-
Reactive oxygen species
- GISTs:
-
Gastrointestinal stromal tumors
References
Smit DL, Mensenkamp AR, Badeloe S, Breuning MH, Simon MEH, van Spaendonck KY et al (2011) Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 79:49–59
Schmidt LS, Linehan WM (2014) Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis 7:253–260
Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML et al (2003) Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73:95–106
De Velasco G, Muñoz C, Sepúlveda JM, Castellano D (2015) Sequential treatments in hereditary leiomyomatosis and renal cell carcinoma (HLRCC): case report and review of the literature. Can Urol Assoc J 9:3–4
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D et al (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410
Linehan WM, Rouault TA (2013) Molecular pathways: fumarate hydratase-deficient kidney cancer—targeting the Warburg effect in Cancer. Clin Cancer Res 19(13):3345–3352
Bardella C, El-Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E et al (2011) Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 225:4–11
Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H et al (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20:524–537
Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP et al (2003) Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12:1241–1252
Menko FH, Maher ER, Schmidt LS, Middelton LA, Aittomaki K, Tomlinson I et al (2014) Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 13:637–644
Alrashdi I, Levine S, Paterson J, Saxena R, Patel SR et al (2010) Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Fam Cancer 9:239–243
Committe on Bioethics, Committee on Genetics, the American College of Medical Genetics and Genomics Social, Ethical and Legal Issues Committee (2013) Ethical and policy issues in genetic testing and screening of children. Pediatrics. doi:10.1542/peds.2012-3680
Alam NA, Barclay E, Rowan AJ, Tyrer JP, Calonje E, Manek S et al (2005) Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141:199–206
Wei MH, Toure O, Glenn GM, Pithukpakorn M, Neckers L, Stolle C et al (2006) Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43:18–27
Vahteristo P, Koski TA, Naatsaari L, Kiuru M, Karhu A, Herva R (2010) No evidence for a genetic modifier for renal cell cancer risk in HLRCC syndrome. Fam Cancer 9:245–251
Park H, Kim CS (2015) Natural 10-year history of simple renal cysts. Korean J Urol 56:351–356
Lamba M, Verma S, Prokopetz R, Pierscianowski TA, Jabi M, Moyana T (2005) Multiple cutaneous and uterine leiomyomas associated with gastric GIST. J Cutan Med Surg 9:332–335
Matyakhina L, Freedman RJ, Bourdeau I, Wei MH, Stergiopoulos SG, Chidakel A et al (2005) Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90:3773–3779
Lehtonen HJ, Kiuru M, Ylisaukko-oja SK, Salovaara R, Herva R, Koivisto PA et al (2006) Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43:523–526
Carvajal-Carmona LG, Alam NA, Pollard PJ, Jones AM, Barclay E, Wortham N et al (2006) Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab 91:3071–3075
Acknowledgments
The authors would like to thank the patients for their willingness to participate. We are grateful to Jorge Eduardo Caminos Pinzon, PhD in neuroendocrinology and member of the Department of Physiological Sciences at the Faculty of Medicine, Universidad Nacional de Colombia, for kindly letting us use one of his genetics labs, and for all his assistance. Also to Maria Fernanda Garces Gutierrez, MSc Human Genetics and PhD student of Biotechnology, Universidad Nacional de Colombia for all her help in the lab. Finally, we want to thank Alvaro Sierra, who helped us with the blood extraction.
Authors’ contribution
CAV and CAD conceived the study, participated in its design and coordination, and in writing the manuscript. CAV, CAD, MRL, collected clinical data. CAV, CAD, EGR interpreted all clinical data. ACB and CAV participated in the genetic analysis. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Arenas Valencia, C., Rodríguez López, M.L., Cardona Barreto, A.Y. et al. Hereditary leiomyomatosis and renal cell cancer syndrome: identification and clinical characterization of a novel mutation in the FH gene in a Colombian family. Familial Cancer 16, 117–122 (2017). https://doi.org/10.1007/s10689-016-9922-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-016-9922-4