
Abstract
Non-heading Chinese cabbage (Brassica campestris L. ssp. chinensis Makino), an important vegetable in East Asian countries, is rich in vitamin C, dietary fiber, and other health-enhancing factors. As a staple vegetable, its genetic studies have been hindered due to the lack of polymorphic genetic markers. In this research, 2.3 and 2.1 Gb RNA-seq data have been obtained for a heat-sensitive cultivar “GHA” and a heat-resistant cultivar “XK”, respectively. Totally, 3267 novel transcripts were identified from the assembled 29,037 transcripts. A total of 139 differential expressed genes (DEGs) have been identified. Most DEGs were involved in the signal transduction and general function prediction. Moreover, 37 polymorphic EST-SSR loci and 285,573 SNP markers have been developed. This study will provide valuable transcript sequences and functional markers for further genetic diversity analysis, quantitative trait locus identification, as well as molecular marker assisted selection of Brassicaceae species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassica campestris L. ssp. chinensis Makino (Packoi), also known as non-heading Chinese cabbage (chromosomal group AA, 2n = 2x = 20), is a member of the Brassicaceae family. As a most economically important leafy vegetable in East Asia, non-heading Chinese cabbage is a valuable source of vitamin C, dietary fiber, and other health-enhancing factors (Cheng et al. 2009; Liu et al. 2014a). High temperature is the major limiting factor in vegetable production (Laurie and Stewart 2006; Sánchez et al. 2014). Non-heading Chinese cabbage can’t well grow at high temperatures (exceeding 25 °C). Therefore, the identification and isolation of thermo-tolerance genes and linked markers are very important. However, the publicly available data is limited for the elucidation of molecular mechanisms and gene expression for regulation of heat tolerance cabbage. Moreover, the genetic research of non-heading Chinese cabbage has also been hindered due to lack of efficient genetic markers.
Microsatellites or simple sequence repeats (SSRs), containing genomic-SSRs and expressed sequence tags (EST)-SSRs, are useful for genetic diversity and population structure analysis, as well as marker-assisted breeding, due to the co-dominant inheritance, dispersal throughout the whole genome, polymorphism, abundance, locus-specificity, and high reproducibility (Powell et al. 1996; Wang et al. 2010). In addition, single-nucleotide polymorphisms (SNPs), identified through the comparisons of genome or transcriptome sequences, are also useful in constructing high-resolution genetic maps, investigating population evolutionary history, analyzing genetic diversity, and verifying marker-trait linkages (Chopra et al. 2015).
In this study, a large expressed sequence dataset based on RNA-seq was generated from two non-heading Chinese cabbage cultivars with different heat tolerances. Besides the identification of genes conferring heat-stable resistance, microsatellite and SNP markers have also been developed. This research may lay a theoretical basis for breeding of heat-resistant non-heading Chinese cabbage cultivars.
Materials and methods
Plant materials
Sterile seeds of non-heading Chinese cabbage cultivars (2n = 2x = 20), containing both heat-sensitive cultivar “GHA” and heat-resistant cultivar “XK”, were sown in pots and germinated in a growth chamber under the condition of 20 °C and 75% relative humidity. Furthermore, seedlings of the two varieties grew up under controlled conditions (25 °C, 75% relative humidity) in the growth chamber. For heat stress treatment, the three-week-old seedlings (five-leaves stage) were grown at 37 °C high temperature for 24 h. Then, tender leaves were collected from five plants and pooled for total RNA extraction and transcriptome sequencing. In addition, 36 accessions with morphological differences were collected for validation of microsatellite markers (Supplementary file 1). All these materials were conserved at the Wuhan Vegetable Research Institute (Wuhan, Hubei, China).
Total RNA extraction and transcriptome sequencing
Total RNA was extracted from tender leaves with the TRIzol kit (Takara) according to the manufacturer’s instructions. The mRNA was enriched with magnetic beads coated with oligo (dT)n, and then fragmented, which was reverse transcribed to first-stranded cDNA. Double-stranded cDNA was synthesized with random hexamer primers, purified and added with sequencing linkers. Fragments of the correct size were purified with Universal DNA Purification Kit, and the sequencing libraries were prepared with the RNA-Seq Library Construction Kit. The quality and quantity of the libraries were verified using an Agilent 2100 Bioanalyzer and ABI real time RT-PCR. The qualified cDNA libraries were used for paired-end sequencing on an Illumina HiSeq 2000 platform. The sequencing raw data was deposited in a SRA database at the NCBI website.
Data processing and annotation
Raw sequence reads were filtered by the Illumina pipeline. Then the clean reads were subjected to the TopHat2-Cufflinks-Cuffmerge-Cuffdiff standard pipeline for identification of differentially expressed genes (DEGs). These DEGs were blasted against the Gene Ontology database (GO), Kyoto encyclopedia of Genes and Genomes database (KEGG), and Cluster of Orthologous Groups of proteins database (COG) for enrichment analysis.
Development of polymorphic microsatellite markers
The high quantity filtered transcriptome reads were obtained by Illumina HiSeq 2000 sequencing. Contigs were assembled with Trinity tools (Grabherr et al. 2011). Then, non-redundant unigenes were created from paired-end reads and used for the development of EST-SSR markers with MicroSAtellite (MISA, http://pgrc.ipk-gatersleben.de/misa). The settings for minimum number of repeats were 10, 6, 5, 5, 4, and 4 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs, respectively. For compound SSRs, the maximum distance between the two SSRs was 50 bp. Within these SSR-containing sequences, unique microsatellites with sufficient flanking regions were chosen for primer pair design using the online software Primer 3 (Wang et al. 2010).
Polymorphism of these EST-SSR primers was assessed in 36 individuals of non-heading Chinese cabbage. DNA was extracted from fresh leaves with a modified CTAB (cetyltrimethyl ammonium bromide) method, and quantified by agarose gel electrophoresis. After optimizing amplification conditions for each primer pair, the PCR amplification was performed in a volume of 15 μL consisting of 1.5 μL 10× PCR buffer, 0.3 μM for each primer, 50 ng genomic DNA, 250 μM each dNTP, as well as 0.5 U Taq DNA polymerase (TianGen, Beijing, China). The PCR amplification conditions were set to include initial denaturation at 95 °C for 10 min, followed by 35 cycles (94 °C for 30 s, annealing for 30 sat the optimal temperature listed in Table 1, and 72 °C for 40 s), and a final 10 min elongation step at 72 °C. PCR products were separated on 6% (w/v) denaturing polyacrylamide sequencing gels and visualized by silver staining. The size of DNA fragments was determined by comparison to a 20 bp DNA ladder marker of 20–600 bp (Tiangen, Beijing, China) (Pan et al. 2007).
Data analysis
PCR products were scored manually, and a 0/1 binary matrix was set according to the presence and absence of corresponding amplified bands. Parameters of genetic diversity including number of alleles (Na) per locus, expected heterozygosity (H E ), observed heterozygosity (H O ), tests for linkage disequilibrium (LD), and deviation from Hardy–Weinberg equilibrium (HWE) were calculated with the ARLEQUIN 3.01 software (Wang et al. 2010). In addition, the occurrence of null allele frequencies (NAF) was also estimated by MICROCHECKER 2.2.3 software (van Oosterhaut et al. 2004).
Results
RNA-seq of two non-heading Chinese cabbage cultivars
After stringent quality assessment and data filtering, a total of 2.3 and 2.1 Gb reads were produced from cultivar “GHA” and cultivar “XK” cDNA libraries, respectively (Table 1). The raw data was deposited in a SRA database (NCBI) under the accession number SRP064703 (“XK”: SRR5169348; “GHA”: SRR5169349). After filtering the low quality sequences and trimming low quality bases, 23,519,534 and 29,253,759 reads were aligned to the reference genome (http://brassicadb.org/brad/downloadOverview.php) for cultivars “GHA” and cultivar “XK”, respectively. In particular, 13,116,870 and 18,288,026 unique reads were mapped for “GHA” and “XK”, taking up 55.8 and 62.5%, respectively.
In total, 96.8% of the reads were in intragenic regions, while intergenic reads made only 3.2% in the heat-sensitive cultivar “GHA”. Similarly in the heat-resistant cultivar “XK”, percentages of reads located in intragenic and intergenic regions were 96.9% and 3.1%, respectively. As expected, most reads (92.8% for “GHA” and 92.7% for “XK”) were distributed in the exon regions. In contrast, percentage of reads located in intronic regions was only 4% and 4.2% for cultivar “GHA” and cultivar “XK”, respectively. After optimization of both the start and stop sites, a total of 29,037 non-redundant unigenes were assembled (“GHA”: 23,794, “XK”: 24,524). To our excitement, 3736 novel transcripts were obtained, which produced 3267 novel gene.
Totally, 139 genes were considered to be differentially expressed between these two cultivars. Compared with “GHA”, the expression levels of 87 DEGs were higher in “XK”, while the expression levels of the other 52 DEGs were lower (Fig. 1). NAC domain-containing proteins, MYB protein family, heat shock proteins, ATP binding proteins, superoxide dismutase, as well as other hormone-responsive element, were main members of these DEGs, inferring that these genes might conferred heat tolerance to non-heading Chinese cabbage. These DEGs can be categorized into three main divisions: biologcial processes, cellular components, and molecular functions (Fig. 2). Among the 26 COG categories, signal transduction mechanisms was the largest group, general function prediction only was the second group, while the third group was translation, ribosomal structure and biogenesis (Fig. 3).

A pie chart showing the fraction of genes conferring heat tolerance traits in non-heading Chinese cabbage. (Color figure online)

Histogram presentation of GO classification of genes from B. rapa transcriptome sequences. The blue bars and red bars referred to DEGs and all unigenes, respectively. (Color figure online)

COG function classification of consensus sequences (x axis represented the 26 different groups, y axis referred to the percentage of genes). (Color figure online)
Characteristics and development of EST-SSR markers
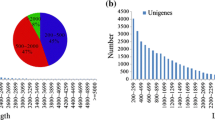
Using a perl script known as MISA (Wang et al. 2013), 19,522 SSR loci were found in 14,653 sequences. A set of 3627 sequences contained more than one EST-SSR loci. Totally, 2105 compound SSRs were identified. Mononucleotide repeat was the main type, with a frequency of 58.124% (11,347), followed by trinucleotide (22.211%, 4336), dinucleotide (19.02%, 3714), tetranucleotide (0.482%, 94), pentanucleotide (0.082%, 16), and hexanucleotide repeats (0.077%, 15) (Fig. 4).

Frequency distribution of different EST-SSRs in non-heading Chinese cabbage (a repeat types; b repeat number)
The most mononucleotide repeat motif was A/T (55.916%). Among the dinucleotide repeat motifs, AG/CT, AT/TA, and AC/GT were the most abundant with frequencies of 14.9%, 2.23% and 1.87%, respectively, while the CG/GC repeat only made 0.026%. For trinucleotides, AAG/CTT, AGG/CCT, and ATC/GAT were the most common types, with the frequencies of 7.535%, 3.55%, and 3.05%, respectively. Repetitions of SSR loci ranged from 5 to 25, and EST-SSRs with ten repeats were the most abundant, followed by those with five, six, and eleven random repeats (Fig. 4).
From the 19,522 primer pairs, 80 were randomly chosen for further validation (Supplementary file 2). Among these EST-SSR primer pairs, 44 produced specific amplification products, while the other 36 primers could not amplify the target products even when annealing temperature was reduced by 10 °C. In particular, 37 primers were polymorphic, with a polymorphic proportion of 46.25%.
These 37 primer pairs were used for genetic diversity analysis in 36 accessions (Table 2). A total of 104 alleles were obtained, with an average of 2.81 alleles per locus (Table 2). However, CaSSR-26, CaSSR-32, and CaSSR-51 all gave out five alleles. The observed Heterozygosity (H O ) and expected Heterozygosity (H E ) ranged from 0.0000 to 1.0000 and 0.0881 to 0.7738, respectively (Table 3). The average H O and H E were 0.4452 and 0.4354, respectively. However, 17 EST-SSR loci significantly deviated from Hardy–Weinberg equilibrium (Table 3), which might be due to the directional selection towards importantly economic traits. Further analysis, conducted with MICROCHECKER software (van Oosterhaut et al. 2004), showed that null alleles existed in these microsatellite loci. Moreover, no significant linkage disequilibrium (LD) was found among these polymorphic EST-SSR loci after Bonferroni correction (Rice 1989).
Detection of SNP markers
Totally, 285,573 putative SNPs have been identified, and 247,708 were heterozygous, while 37,865 were the homozygous SNPs. Moreover, numbers of the synonymous, missense, stopgain, and stoploss were 162,900, 65,321, 284, and 42, respectively. A set of 228,546 SNPs were found in exon regions, and 62 SNPs were in splicing regions. However, no SNPs were found in sequences coding NcRNA, 5′UTR, and even 3′UTR. In addition, 6053 and 9168 SNPs were in intronic and intergenic regions, respectively. At the same time, 15,021 and 21,295 SNPs were present in upstream or downstream flanking regions. Among these SNP loci, the C:G → T:A mutation had the highest rate, followed by T:A → C:G and T:A → A:T mutation types (Fig. 5).

Distribution of mutation types. (Color figure online)
A total of 23,788 heterozygous and 5079 homozygous InDel markers were found. Among them, 1813 InDels were frameshift insertions, and 2303 InDel were non-frameshift insertions. Simultaneously, numbers of InDels corresponding to frameshift deletion and non-frameshift deletion were 1976 and 2430, respectively. No frameshift block substitutions or non-frameshift block substitution InDel were found. The numbers of stopgain and stoploss InDels were 111 and 34, respectively. A total of 8667 InDels were located in exon regions, and 44 InDels were found in splicing regions. Additionally, 14 InDels were found in 5′UTR sequences. Numbers of InDels located in intronic, upstream, downstream, and intergenic regions were 1469, 6426, 8496 and 1217, respectively. InDels with a length of 1nt were the most frequent type, while 2–3nt InDels types followed. In particular, InDels with lengths more than 10nt were very rare.
Discussion
With the advent and rapid development of next generation sequencing (NGS) technologies, RNA sequencing (RNA-seq) shows to be powerful and reliable tools for gene expression profile analysis, identification of candidate genes for target traits, genetic map construction, and development of molecular markers in many species including the genus Brassica (Shendure and Ji 2008; Kucuktas et al. 2009; Manuel et al. 2011; Liu et al. 2014b; Xiao et al. 2015). Recently, a large number of genomic and transcriptomic data has been made available for both model and non-model organisms, including Oryza sativa, Arabidopsis, cucumber, as well as Chinese cabbage. These high-throughput data is helpful in understanding the complexity of plant growth, development, and responses of plants to environmental stress. However, little data for heat stress in non-heading Chinese cabbage has been reported to our knowledge. To identify genes conferring heat tolerance and isolate SSR markers linked with these genes in non-heading Chinese cabbage, a heat-sensitive cultivar “GHA” and a heat-resistant cultivar “XK” were chosen for transcriptome sequencing.
The BcHSP81-4 gene, a member of heat shock proteins, was identified from a suppression subtractive hybridization cDNA library in non-heading Chinese cabbage (Brassica campestris ssp. chinensis Makino), which is also responsive to salt stress and cold stress (Liu et al. 2011). In this study, heat shock proteins have also been found to be involved in heat tolerance. In addition, NAC domain-containing proteins, MYB protein family, and hormone-responsive elements were also important gene products in response to heat stress, which were in agreement with reports in soybean (Irsigler et al. 2007), citrus (Oliveira et al. 2011), and rice (Fang et al. 2015).
In related to genomic-SSRs identified from random genomic sequences, EST-SSR markers are potentially more efficient for gene targeting, QTL mapping, and marker-assisted breeding due to their potentially linkage with particular transcriptional regions contributing to agronomic phenotypes (Bozhko et al. 2003; Zheng et al. 2013; Scott et al. 2000). The distribution density of EST-SSR markers in non-heading Chinese cabbage (one loci per 2.37 kb) was higher than that of other reported species, such as rice (one SSR loci per 3.4 kb), Amorphophallus (one SSR loci per 3.63 kb), wheat (one SSR loci per 5.4 kb), soybean (one SSR loci per 7.4 kb), Arabidopsis (one SSR loci per 14 kb), and even one loci per 20 kb in cotton (Zheng et al. 2013; Peng and Lapitan 2005; Varshney et al. 2002). The small genome size might contributed a lot to the high frequency of EST-SSR loci in non-heading Chinese cabbage.
The trinucleotide repeat motif (22.21%) was more frequent than dinucleotide type (19.02%), which was consistent with the EST-SSR distributions reported in radish (Wang et al. 2012). However, dinucleotide repeat was the most abundant in spruce (Rungis et al. 2004), Cucurbita pepo (Gong et al. 2008), Momordica charantia (Wang et al. 2010), pigeonpea (Dutta et al. 2011), as well as Amorphophallus (Zheng et al. 2013). Moreover, the AG/CT repeat motif was the most common dinucleotide type, which was in agreement with most plant species (Pan et al. 2007; Wang et al. 2010). Most of these SSR loci have two alleles, suggesting the relatively low polymorphism in non-heading Chinese cabbage.
As a virtually unlimited, bi-allelic, evenly distributed along genome, and co-dominant resource, SNPs are highly valuable for research and modern breeding. Until now, only a few SNP markers have been described in non-heading Chinese cabbage. Rahman et al. have developed 24 SNPs with more than 2-kb sequence for the major seed coat color gene in Brassica rapa (Rahman et al. 2007). Moreover, 151 SNP markers have been developed from B. rapa, which was used to construct the linkage map (Li and Hinaba 2009). Chung et al. have also constructed a genetic map with SNP markers and mapped a TuMV resistance locus in B. rapa (Chung et al. 2014).
The EST-SSRs and SNPs characterized in this study possessed important implications for genetics study and molecular breeding in non-heading Chinese cabbage. Moreover, these markers will be useful for constructing high-density genetic linkage maps, mapping quantitative trait loci, assessing germplasm polymorphism and evolution, marker assisted selection, and cloning functional gene in Chinese cabbage.
References
Bozhko M, Riegel R, Schubert R, Müller-Starck G (2003) A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol Ecol 12(11):3147–3155
Cheng Y, Geng J, Zhang J, Wang Q, Ban Q, Hou X (2009) The construction of a genetic linkage map of non-heading chinese cabbage (Brassica campestris ssp. chinensis makino). J Genet Genomics 08(8):501–508
Chopra R, Burow G, Farmer A, Mudge J, Simpson CE, Wilkins TA, Baring MR, Puppala N, Chamberlin KD, Burow MD (2015) Next-generation transcriptome sequencing, SNP discovery and validation in four market classes of peanut, Arachis hypogaea. Mol Genet Genomics 290:1169–1180
Chung H, Jeong YM, Mun JH, Lee SS, Chung WH, Yu HJ (2014) Construction of a genetic map based on high-throughput SNP genotyping and genetic mapping of a tumv resistance locus in Brassica rapa. Mol Genet Genomics 289(2):149–160
Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, Dogra V, Gaikwad K, Sharma TR, Raje RS, Raje RS, Bandhopadhya TK (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol 11(1):17
Fang Y, Liao K, Du H, Xu Y, Song H, Li X, Xiong L (2015) A stress-responsive NAC transcription factor SNAC3 confers heat and drought tolerance through modulation of reactive oxygen species in rice. J Exp Bot 66(21):6803–6817
Gong L, Stift G, Kofler R, Pachner M, Lelley T (2008) Microsatellites for the genus Cucurbita and an SSR-based genetic linkage map of Cucurbita pepo L. Theor Appl Genet 117(1):37–48
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Irsigler AS, Costa MD, Zhang P, Reis PA, Dewey RE, Boston RS, Fontes EPB (2007) Expression profiling on soybean leaves reveals integration of er- and osmotic-stress pathways. BMC Genomics 8(4):431
Kucuktas H, Wang S, Li P, He C, Xu P, Sha Z, Liu H, Jiang Y, Baoprasertkul P, Somridhivej B, Wang Y, Abernathy J, Guo X, Liu L, Muir W, Liu Z (2009) Construction of genetic linkage maps and comparative genome analysis of catfish using gene-associated markers. Genetics 181(4):1649–1660
Laurie S, Stewart GR (2006) Effects of nitrogen supply and high temperature on the growth and physiology of the chickpea. Plant Cell Environ 16(6):609–621
Li F, Hinaba K (2009) A Brassica rapa linkage map of EST-based SNP markers for identification of candidate genes controlling flowering time and leaf morphological traits. DNA Res 16(6):311–323
Liu T, Hou X, Zhang J, Song Y, Zhang S, Li Y (2011) A cdna clone of bchsp81-4 from the sterility line (pol cms) of non-heading Chinese cabbage (Brassica campestris ssp. chinensis). Plant Mol Biol Rep 29(3):723–732
Liu T, Dai W, Sun F, Yang X, Xiong A, Hou X (2014a) Cloning and characterization of the nitrate transporter gene branrt2.1 in non-heading Chinese cabbage. Acta Physiol Plant 36(4):815–823
Liu F, Sun F, Xia JH, Li J, Fu GH, Lin G, Tu RJ, Wan ZY, Quek D, Yue GH (2014b) A genome scan revealed significant associations of growth traits with a major QTL and GHR2 in tilapia. Sci Rep 4:7256
Manuel G, Grabherr MG, Mitchell G, Cole T (2011) Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 8(6):469–477
Oliveira TMD, Cidade LC, Gesteira AS, Filho MAC, Filho WSS, Costa MGC (2011) Analysis of the NAC transcription factor gene family in citrus reveals a novel member involved in multiple abiotic stress responses. Tree Genet Genomes 7(6):1123–1134
Pan L, Quan Z, Li S, Liu H, Huang X, Ke W, Ding Y (2007) Isolation and characterization of microsatellite markers in the sacred lotus (Nelumbo nucifera gaertn.). Mol Ecol Notes 7(6):1054–1056
Peng J, Lapitan NLV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomic 5(2):80–96
Powell W, Machray GC, Provan J (1996) Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1(7):215–222
Rahman M, Mcvetty PBE, Li G (2007) Development of SRAP, SNP and multiplexed SCAR molecular markers for the major seed coat color gene in Brassica rapa. Theor Appl Genet 115(8):1101–1107
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rungis D, Berube Y, Zhang J, Ralph S, Ritland CE, Ellis BE, Douglas C, Bohlmann J, Ritland K (2004) Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor Appl Genet 109(6):1283–1294
Sánchez C, Baranda AB, Marañón IMD (2014) The effect of high pressure and high temperature processing on carotenoids and chlorophylls content in some vegetables. Food Chem 163(20):37–45
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. Theor Appl Genet 100(5):723–726
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145
van Oosterhaut C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7(2A):537–546
Wang S, Pan L, Hu K, Chen C, Ding Y (2010) Development and characterization of polymorphic microsatellite markers in Momordica charantia (Cucurbitaceae). Am J Bot 97(8):e75–e78
Wang S, Wang X, He Q, Liu X, Xu W, Li L, Gao J (2012) Transcriptome analysis of the roots at early and late seedling stages using illumina paired-end sequencing and development of EST-SSR markers in radish. Plant Cell Rep 31(8):1437–1447
Wang H, Jiang J, Chen S, Qi X, Peng H, Li P, Song A, Guan ZY, Fang WM, Liao Y, Chen F (2013) Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 8(4):e62293
Xiao S, Han Z, Wang P, Han F, Liu Y, Li J, Wang ZY (2015) Functional marker detection and analysis on a comprehensive transcriptome of large yellow croaker by next generation sequencing. PLoS ONE 10(4):e0124432
Zheng X, Cheng P, Ying D, You Y, Yang C, Hu Z (2013) Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genomics 14(2):414–416
Acknowledgements
This work was supported by Fund of National Commodity Vegetable Technology System (CARS-25-G-30), the Natural Science Foundation of Hubei Province of China (2013CFA102), Hubei Collaborative Innovation Center for the Characteristic Resources Exploitation of Dabie Mountains (2015TD07), and Breeding and application of late bolting Chinese Cabbage (2016000311).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, S., Zhou, G., Huang, X. et al. Transcriptome analysis of non-heading Chinese cabbage under heat stress by RNA-seq and marker identification. Euphytica 213, 109 (2017). https://doi.org/10.1007/s10681-017-1891-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1891-7