
Abstract
Pakchoi is an important vegetable grown in worldwide that prefers a cool climate, and its growth is severely affected by high temperatures in the summer. Little is known for the molecular regulatory mechanisms of high-temperature stress in pakchoi. The limited availability of gene sequence has greatly affected molecular breeding and functional genomic analysis. In this study, we conducted comprehensive analyses for heat treatment in pakchoi by RNA-Seq. Finally, 64.29 million clean reads containing 32,666 unigenes with an N50 length of 1405 bp, and a total length of 33.39 Mb were identified. Overall, 11,024 SSRs located in 8404 unigenes were obtained; these findings will be very useful for molecular assisted breeding. Totally, 1220 differentially expressed genes (DEGs) were detected between the high-temperature group (TH) and the control (CK), among which 699 DEGs were up-regulated and 521 were down-regulated. The enrichment analysis indicated that 12 GO subcategories and 9 KEGG pathways were enriched significantly (P value <0.05). Finally, six DEGs were validated by qRT-PCR, and the results further verified the reliability of RNA-Seq. Herein, we present the first comprehensive characterization of heat-treated pakchoi using transcriptome analysis. Several important genes that respond to high temperature were identified, and their roles in heat stress responses are discussed. In conclusion, our study represents a fully characterised pakchoi transcriptome. In addition, it provides the important resources for future genomic and genetic studies of pakchoi under heat stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pakchoi (Brassica rapa L. chinensis), also called non-heading Chinese cabbage, originated from the middle and lower area of Yangtze River in China. Pakchoi prefers cool weather, which makes it difficult to grow in summer. To meet market demand, it is now cultivated and supplied year-round, and it is one of the major vegetables in China. However, high-temperature stress in summer is the limiting factor for the efficient production of pakchoi. The main symptoms affected by high-temperature stress on pakchoi include leaf scorching, elongation of the dwarf stem, leaf senescence and abscission, loss of vigour, imbalances in photosynthesis and respiration, and reductions of dry mass of shoot, and net assimilation. Therefore, pakchoi production is seriously affected by the high temperatures of summer, especially in southern China. Heat tolerance is a hot topic of research in pakchoi breeding.
Temperature stress affects a serial of biological and molecular functions (Gracey et al. 2004). It has important effects on chemical and physical processes in biological systems (Scott and Johnston 2012). The temperatures stress can affect several properties of structural components and biomolecules of cells, including the activity, folding, stability, and assembly of proteins (Somero 1995); the structure of lipids (Simon et al. 1995; Pan et al. 2008); and the fluidity of cell membranes (Ueda et al. 1976). As these changes absolutely involve the transcriptional regulation of large numbers of genes (Swindell et al. 2007; Zhang et al. 2010), it is important to characterise the specific heat-related pathways and genes that respond to heat stress. The identification of these pathways and genes will enable the mechanisms of heat tolerance to be understood. This new information will enable genetic engineering strategies to be employed to improve heat stress resistance in this vitally important commodity.
RNA-Seq analysis is a mainly phenotyping technique (Mortazavi et al. 2008). Using this method, lots of genes can be identified, and the DEGs between two samples can be uncovered. It is very useful for discovering the relationship between phenotypes and gene expression. The RNA-Seq, using the second-generation sequencers, is a useful tool for detecting the whole-genome gene expression. With its high resolution, RNA-Seq provides detailed information on the gene structure (Van Verk et al. 2013). It has been successfully used to identify potential resistance genes of cold tolerance in olive flounder (Hu et al. 2014) and wheat (Gulick et al. 2005), salt tolerance in the common bean (Hiz et al. 2014), and drought tolerance in cotton (Bowman et al. 2013) and rice (Oono et al. 2014).
The purpose of our study was to use RNA-Seq to investigate the differences in the transcriptional responses of pakchoi grown at a high temperature versus at a suitable temperature. Moreover, we sought to identify those genes that have important roles in heat tolerance. The results will enable a well understanding of transcriptional basis of adaptive heat stress mechanisms in this commodity.
Materials and methods
Materials preparation and stress treatments
The pakchoi heat-resistant inbred line ‘GHQ’ was supplied by the Jiangsu Academy of Agricultural Sciences (JAAS), China, for use in this study. The seedlings were grown in trays containing Pindstrup plus peat substrate (Pindstrup Mosebrug A/S, Denmark). The trays were placed in an artificial climate box with 14 h light under 28 °C and 10 h dark under 20 °C. Three-week-old seedlings were exposed to high temperatures (38/30 °C during the day/night). For RNA-Seq, leaves were collected after the seedlings were treated with high temperatures for 120 h (TH). The leaves of seedlings grown at normal temperatures (28/20 °C, day/night) were simultaneously collected as the controls (CK). For qRT-PCR verification, leaves were collected after the seedlings were treated with high temperature for 0, 6, 12, 24, 48, 72, 96, and 120 h, respectively. Each sample harvested randomly from 3 to 4 seedlings and was frozen in liquid nitrogen and stored at −80 °C for RNA isolation.
RNA isolation, library construction, and sequencing
RNA was extracted from the materials using Trizol Reagent according to the manufacturer’s instructions (Invitrogen, CA, USA). The RNA integrity and quality were detected using the Agilent 2100 Bioanalyser (Agilent, USA), and NanoDrop spectrophotometer (NanoDrop, USA), respectively. Subsequently, the mRNA was divided into small fragments under elevated temperature. The paired-end libraries were constructed using Illumina genomic sample prep kit. The cDNA was purified using the Qiaquick PCR Extraction Kit (Qiagen, USA) and then linked with adapters (Margulies et al. 2005). The multiplexed DNA libraries were mixed in equal volumes and normalised to a concentration of 10 nM. Two normalised cDNA libraries were constructed using RNA from the high-temperature treatment and control. The libraries were sequenced by using Illumina Hiseq 2000 platform (Shanghai Personal Biotechnology Co. Ltd, China).
Data filtering and de novo assembly
For the analyses in this study, we constructed a flowchart that started with data filtering and progressed to functional enrichment analyses (Figure S1). The paired-end raw reads were generated with lengths of 101 bp using RNA-Seq. First, the read adaptor contamination was removed from reads, then reads were checked using 5 bp windows from 3′ to 5′ end to trim the bases with the low quality (Q < 20). The reads with final lengths of less than 50 bp were also removed. The sequences with low quality (Q < 5) and ambiguous ‘N’ bases (‘N’ ratio >10 %) were removed from the raw reads. After filtering, the reads were assembled into contigs, transcripts, and unigenes by the Trinity Software (http://trinityrnaseq.sf.net) with the default settings, except for K-mer value. All genes were searched against the non-redundant database of the NCBI using BLAST program, and the best-hit transcripts were chose as unigenes.
The gene annotation, gene expression, and functional enrichment analyses
All assembled unigenes were annotated by comparing their sequences with the publicly available protein databases, including the Non-redundant (http://www.ncbi.nlm.nih.gov), KEGG (http://www.genome.jp/kegg), GO (http://www.geneontology.org), and evolutionary genealogy of genes: NOGs (http://eggnog.embl.de) databases by BLAST (e value <1 × 10−5). The RPKM defined as reads per kilobase of exon per million mapped reads were used to normalise the abundances of genes (Mortazavi et al. 2008). The P value was calculated using DEseq program (Anders and Huber 2010). Twofold change (FC > 2) and P value <0.05 were used to define the significantly differentially expressed genes (DEGs) between treatment and control. The ‘volcano plot’ indicated the distribution of DEGs. The pathways involving the DEGs were detected by the KEGG database, and the KEGG mapper (http://www.genome.jp/kegg/tool/map_pathway2.html) was used to show the up-regulated and down-regulated genes. The GO enrichment analyses were performed by Blast2GO (Conesa et al. 2005; Gotz et al. 2008). We also conducted enrichment analyses using the KEGG database.
The SSR markers mining for pakchoi
The Microsatellite identification software (http://pgrc.ipk-gatersleben.de/misa/) was used to detect SSR markers. The threshold for mono-, di-, tri-, tetra-, penta-, and hexa- nucleotide repeat motifs was set at 10, 6, 5, 5, 5, and 5, respectively. The maximal number of bases interrupting 2 SSRs in a compound microsatellite was set as 100 according to a previous report (Song et al. 2015).
Validation of DEGs expression with quantitative real-time PCR
The six randomly selected DEGs were quantified and verified by quantitative real-time PCR (qRT-PCR). The primers were designed according to the sequencing data by Primer 5.0 (Table S1). The RNA was got from the samples treated at 38/30 °C (day/night) after 0, 6, 12, 24, 48, 72, 96, and 120 h. Reverse cDNA was also synthesised using PrimeScriptTM RT Reagent Kit (Takara). The qRT-PCR was conducted by Eppendorf Mastercycler ep realplex. The actin gene of pakchoi was used as an internal control to normalise the expression levels, and all experiments were conducted in triplicate (Xu et al. 2013). The reaction was performed in a total 20 μL volume, which contained 10 μL 2× SYBR real-time PCR premixture (Takara), 1 μL diluted cDNA mix, and 0.4 μL of each primer (10 mM). The thermal profile for the SYBR Green RT-PCR was set as 95 °C for 4 min, then 35 cycles of 95 °C for 15 s, 57 °C for 15 s, and 72 °C for 25 s. The expression levels of each gene were analysed by the comparative CT method (2−ΔΔCT method) (Livak and Schmittgen 2001).
Results
RNA-Seq and de novo assembly of pakchoi transcriptome
To obtain an overview of the pakchoi transcriptome under high-temperature treatment, we constructed and sequenced two libraries from the heat-treated (TH) and control (CK) groups. A large number of datasets were generated by Illumina HiSeq 2000 when the two pakchoi cDNA libraries (TH and CK) were sequenced. According to the Illumina RNA transcriptome sequencing, a sequence can generate 2 × 101 base pairs (bp) from each paired-end (PE) of a cDNA fragment. In total, there were 40,879,145 and 23,413,471 raw reads in the TH and CK groups, respectively (Table 1). We used stringent criteria for quality control by removing the low- quality reads. After filtering, the base quality of the reads was checked and analysed using FastQC. The results showed that the quality of bases in the middle of the reads was larger than that of the two ends of the reads (Figure S2a). However, the quality of most bases was larger than 32 (Q > 32), and the quality of most reads was approximately 37 (Figure S2a, b). In addition, we checked the percentages of A, T, C, and G in each clean read. The results showed that these percentages in the middle and 3′ ends were relatively stable, and there were only fluctuations at the 5′ ends (Figure S3a). Furthermore, the curve of the measured mean GC content was highly consistent with the expected value (Figure S3b). Therefore, the clean reads were of high quality and could be used for further assembly and analyses. In total, 34,803,854 (TH) and 19,635,919 (CK) high-quality, clean paired-end sequencing reads with a total of 6721,791,428 and 3,795,416,203 nucleotides were obtained for the two libraries, respectively (Table 1).
The two libraries reads were combined to assemble the pakchoi transcriptome. After the first assembly step, a total of 169,324 contigs were obtained with a total length over 58.01 Mb (Table 2). The contig size was 343 bp averagely, and contig N50 was 616 bp. Most contigs (61 %) were less than 200 bp, while only 14 % of the contigs were more than 500 bp (Figure S4). The contigs were joined into 94,808 genes with length of 851 bp and total length of 80.68 Mb (Table 2). Forty-seven percent of the transcripts were between 500 and 2000 bp in length, and 8 % of the transcripts had lengths over 2000 bp (Fig. 1a). Finally, 32,666 unigenes got after combining the transcripts, with the length of more than 33.39 Mb. The unigene length was 1022 bp averagely, and N50 was 1405 bp (Table 2). Sixty percent of the unigenes were between 500 and 2000 bp in length, and 10 % of the unigenes were larger than 2000 bp (Fig. 1b). Therefore, the quality of this assembled unigenes was credible and accurate.

Length distributions and statistical analyses. a Transcripts; b unigenes
Classification and function annotation of the unigenes in pakchoi
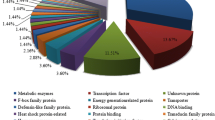
All 32,666 unigenes were annotated according to Nr, KEGG, GO, and eggNOG databases using the BLAST program (E value <10−5). Most genes significantly matched in at least one of these databases. For example, the eggNOG analysis revealed 25,946 unigenes with significant homology, which were then assigned to the appropriate eggNOG functional categories. The eggNOG database is used to divide the genes into several categories based on the protein function and the domain. The eggNOG annotation results showed that the unigenes were classified into 26 functional categories (Figure S5; Table S2). However, most unigenes belonged to the “function unknown” and “general function prediction only” categories (26.49 and 15.79 %, respectively), followed by “signal transduction mechanisms” (8.05 %), “post translational modification, protein turnover, and chaperones” (7.44 %), and “transcription” (5.10 %). These annotations will provide rich valuable resources for studying the specific processes and pathways involved in heat stress in pakchoi or other related species.
Identification of SSR loci and analysis of their characteristic information
The transcriptome datasets are very important resources for the rapid development of genetic markers. A total of 11,024 SSRs were detected, which were located in 8404 unigenes; 2021 genes contained more than 1 SSR (Table S3). The largest fraction of SSRs identified was mononucleotide (4603), followed by tri-nucleotide (3175), and di-nucleotide (3163) (Fig. 2a, b; Table S4). Among the mononucleotide SSRs, A/T (4564) accounted for 99.2 % of the mononucleotide repeats, while only contained 39 C/G repeat. In the di-nucleotide SSRs, AG/CT (2628) accounted for 83.1 % of the di-nucleotide repeats, and no CG/CG repeats were detected. In the tri-nucleotide SSRs, AAG/CTT repeats’ number (1124) was greater than that of other tri-nucleotide repeats. The average frequency of SSRs was one SSR per 3.0 kb. All of these SSRs identified in our transcriptome datasets will be very useful resource for the development of markers.

SSR analyses in unigenes of the pakchoi transcriptome. a Number of SSRs for each SSR type; b the summary of SSRs from mono- to pentanucleotide
Detection of differentially expressed genes between heat-treated and control pakchoi
The expression of each gene was calculated by RPKM to normalise the transcript abundances (Table S5). To identify the temperature response genes, 1220 significant DEGs were found between the heat-treated and control groups (FC > 2 and P value < 0.05). Among these unigenes, 699 genes were up-regulated in TH compared with CK (Fig. 3a; Table S6), and 521 genes were down-regulated in TH compared with CK (Table S7). We also conducted a cluster analysis for these DEGs and obtained robust cluster results for the up- and down-regulated genes (Fig. 3b). On the basis of this study, these genes were inferred to be involved in the stress response to high temperatures in pakchoi, and they may have important regulatory roles in the heat resistance for pakchoi.

DEGs analyses between heat-treated and control pakchoi. a Volcano plot for the DEGs analysis. The x-axis represents the log-transformed fold change, and the y-axis represents the -Log10 (P value). The distributions of DEGs (P value ≤0.05 and FC ≥2 or ≤0.5) identified between the heat-treated and control samples. The red dots represent up-regulated genes; the green dots represent down-regulated genes; b hierarchical clustering analysis of DEGs using the expression values
The GO and pathway enrichment analyses revealed the function of key DEGs related to heat stress
To understand the functions of the DEGs, we performed the GO enrichment analyses using all genes as background. According to GO, the significant DEGs were divided into three major functional categories, including biological process, molecular function, and cellular component (Table S8). These three categories could further divided into 54 subcategories, and 12 of them were significantly enriched with the P value <0.05 (Fig. 4; Table 3). Most enriched subcategories belonged to the categories of biological process (8 subcategories) and cellular component (4 subcategories); there was no significant enrichment of subcategories in the molecular function category. Among cellular components, the extracellular region (GO:0005576) was the most enriched subcategory. Among the biological process categories, the four most enriched subcategories were “response to endogenous stimulus (GO:0009719)”, “response to stress (GO:0006950)”, “response to abiotic stimulus (GO:0009628)”, and “response to biotic stimulus (GO:0009607)”. These results showed that most enrichment subcategories were related to stress, abiotic and biotic stimuli, indicating the accuracy of our study. These genes would greatly enhance the potential utilization of pakchoi in heat stress.

GO enrichment analysis of DEGs between heat-treated and control pakchoi. The horizontal dotted line represents the significance level (P value <0.05)
The KEGG database uses molecular-level information for the systematic understanding of high-level gene functions in terms of networks of biological systems, such as cells, organisms, and ecosystems. The unigenes were searched against the KEGG database using BLASTX to identify the biological pathways activated in the leaves of pakchoi in response to heat stress. Of 32,666 annotated unigenes in total, 13,836 unigenes and 489 DEGs had significant matches in this database (Table S9).
All of these DEGs were generally assigned to six categories and were further divided into 38 subcategories (Fig. 5; Table S9). Among 489 DEGs, 352 genes belonged to the metabolism category, followed by organismal systems (51). Interestingly, most significantly enriched DEGs (P value <0.05) belonged to the metabolism category and were assigned into eight subcategories (Fig. 5). The organismal systems’ category only had one significantly enriched subcategory, and the other four categories had no enriched subcategories. In the metabolism category, the first three subcategories were “amino-acid metabolism”, “biosynthesis of other secondary metabolites”, and “xenobiotics biodegradation and metabolism”, which contained 73, 38, and 32 DEGs, respectively. In addition, the “energy metabolism”, “lipid metabolism”, and “environmental adaptation” subcategories were also enriched. All of these enriched pathways and DEGs would be of great use in breeding heat-resistant pakchoi.

KEGG enrichment analysis of DEGs between heat-treated and control pakchoi. The horizontal dotted line represents the significance level (P value <0.05). The histogram at the top-right corner represents the DEG number for each category
Validation of the gene expression using qRT-PCR
To verify that the unigenes obtained from the computational analyses were, indeed, expressed and to analyse the differences in gene expression between the TH and CK groups, qRT-PCR was used to detect the expression of 6 DEGs in pakchoi (Table 4). These six genes included 3 HSPA1_8 proteins (heat-shock 70 kDa protein1/8) as well as HSPBP1 (hsp70-interacting protein), ERF1 (ethylene-responsive transcription factor 1), and PLD (phospholipase D). We detected the expression of these genes after 0, 6, 12, 24, 48, 72, 96, and 120 h of heat treatment. The results indicated that the expression of HSPBP1, ERF1, and PLD was up-regulated rapidly and drastically, and the expression of all three heat-shock 70 kDa genes peaked at 72 h after oscillating between over- and under-expression (Fig. 6). Therefore, most of the qRT-PCR results were consistent with the RNA-Seq data.

Relative expression level of six DEGs among heat-treated and control pakchoi obtained by qRT-PCR. The x-axis indicates the treatment time and the y-axis indicates the relative expression level of genes. Error bars represent standard errors from three independent replicates
Discussion
Feasibility of RNA-Seq and assembly for non-model species with un-sequenced genomes
Understanding the status of transcriptomes is useful for uncovering the complexity of regulation (Mortazavi et al. 2008). In recent years, the second-generation sequencing has enabled the faster creation of large-scale sequence datasets at lower costs compared with the conventional Sanger sequencing. The sequencing lengths can reach 2 × 100 bp with an Illumina HiSeq 2000 system. This is the most accurate and efficient approach to detect the transcriptional boundaries of genes (Fullwood et al. 2009). We obtained transcriptomic data from heat-treated and control pakchoi using Illumina HiSeq 2000 with PE 100 bp sequencing, obtaining a total of ~4 Gb high-quality clean reads for each library.
The length of transcripts is a very important factor that reflects the quality of the data assembly. Compared with the previous studies, the length of transcripts (~851 bp) for pakchoi was shorter than those of T. chinensis (1077 bp) (da Hao et al. 2011) and Jatropha curcas (916 bp) (Natarajan and Parani 2011), but they were longer than those generated from Amaranthus tricolor (831.65 bp) (Liu et al. 2014), Phyllostachys heterocycla (612 bp) (He et al. 2013), S. indicum (629 bp) (Wei et al. 2011), and maize (218 bp) (Xiong et al. 2011). More importantly, 28,558 (30.12 %) of the assembled transcripts in this study were longer than 1000 bp, with an N50 length of 1287 bp (Fig. 1; Table 2).
Among 94,808 transcripts, 32,666 (approximately 34.45 %) were annotated successfully, revealing their relatively conserved functions. However, most genes did not annotate according to the existing databases. This relative scarcity of identifiable genes is also due to the lack of a complete set of genomic or transcriptomic sequences for pakchoi.
Temperature is a highly significant environmental factor during plant development
In this study, RNA-Seq was used to identify the DEGs between heat-treated and control pakchoi. Several genes, intracellular pathways, and key biological processes were involved in high-temperature stress, which likely explain the mechanism of heat tolerance and provide scientific evidence supporting screens for a heat-tolerant germplasm in pakchoi. Some of the DEGs may play important roles in facilitating pakchoi growth under high-temperature conditions.
Role of HSPs in heat tolerance
Most organisms respond to heat stress by generating heat-shock proteins (Walser et al. 2006; Chen et al. 2007; Wray 2007; Kang et al. 2009). Transgenic Arabidopsis containing Trichoderma harzianum Hsp70 is an abiotic, stress tolerant plant (Montero-Barrientos et al. 2010). The proteins of the Hsp70 family are constitutively up-regulated in response to various stressors, such as heat, cold, anoxia, and metal stresses (Li et al. 1999; Sung et al. 2001). The Hsp70 family belongs to the multigene family. For example, E. coli contains three Hsp70 genes and Arabidopsis has 18 Hsp70 family members, of which 14 belong to the Hsp70 family (Lin et al. 2001). In spinach, at least 12 Hsp70 genes have been identified (Guy and Li 1998). In our study, three HSPA genes in pakchoi were all annotated as heat-shock 70 kDa proteins and were up-regulated significantly until 72 h after the high-temperature treatment.
Role of ethylene-responsive transcription factor in heat tolerance
Ethylene response factor (ERF) family proteins comprise a subfamily of the AP2/ERF family and are responsible for generating tolerance to stresses. Thus, a detailed study is required to functionally characterise the roles of specific ERFs in different stresses. ERF genes have been identified as heat-response transcription factors, and an ERF coactivator gene has been shown to be synergistically expressed with ERF under heat stress (Zhang et al. 2012). The expression of AtERF53 in Arabidopsis thaliana has been shown to be induced by heat treatment, reaching a 3.8-fold induction at 4 h (Hsieh et al. 2013). Moreover, ethylene response factor (ERF1, XP_002281052.1) may play a central role in grapevine stress response, because its expression is up-regulated after both heat-and water-deficit stresses (Rocheta et al. 2014). Similarly, reduced anthocyanin accumulation and hastened ripening in response to high temperatures have been associated with the coincidental expression of HSP chaperones and ERF with the repression of transporter-encoding genes (Carbonell-Bejerano et al. 2013). In our study, the expression of ERF1 in pakchoi was up-regulated immediately and dramatically after the high-temperature treatment. Thus, the induction of HSP and the co-activated expression of ERF and Hsf are critical for acclimation to adverse high temperatures.
Role of phospholipase D in heat tolerance
The regulation of heat tolerance is very complicated in species, and several pathways in different cellular compartments are activated. The treatment of high temperature results in increase Ca2+ levels in cells in two ways at the plasma membrane (PM). First, the heat stress can trigger the alteration of membrane fluidity, which leads to the activation of phospholipase D and phosphatidylinositol phosphate kinase (PIPK), as well as the accumulation of phosphatidylinositol 4,5-bisphosphate (PIP2), and phosphatidic acid (PA) (Mishkind et al. 2009). Second, the stress affects the Ca2+ channels opening, which results in increased Ca2+ influx (Saidi et al. 2009). In animal cells, the studies of cation channels involved in heat tolerance also indicate that the putative Ca2+ channels may be proper candidates for PM heat stress sensors (Sokabe et al. 2008; Xiao et al. 2011). Phospholipase D is a critical enzyme involved in membrane phospholipid catabolism response to plant stress. Recently, a novel PLD gene, CbPLD, was cloned and characterised from Chorispora bungeana (Yang et al. 2012). The transcripts of CbPLD were induced and greatly increased in abundance under heat stress. This finding is similar to the results of this study, indicating that PLD also may play an important role in response to high temperatures in pakchoi. The functions and mechanisms of PLD in the acquisition of thermal tolerance in plants remain to be investigated.
In conclusion, this study provides important new insights into heat stress adaptation and should facilitate further studies of pakchoi genes and their functions. This study represents a fully characterised transcriptome and provides rich valuable resources for genomic studies in pakchoi under heat stress.
Author contribution statement
The study was conceived by Hai Xu and Jinfeng Chen. Hai Xu and Xihan Yuan contributed to the bioinformatics analysis. Bo Song and Xiaoxue Fan performed the experiments. Hai Xu and Longzheng Chen prepared and wrote the manuscript. All authors revised the manuscript. All authors read and approved the final manuscript.
References
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Bowman MJ, Park W, Bauer PJ, Udall JA, Page JT, Raney J, Scheffler BE, Jones DC, Campbell BT (2013) RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS One 8:e82634
Carbonell-Bejerano P, Santa Maria E, Torres-Perez R, Royo C, Lijavetzky D, Bravo G, Aguirreolea J, Sanchez-Diaz M, Antolin MC, Martinez-Zapater JM (2013) Thermotolerance responses in ripening berries of Vitis vinifera L. cv Muscat Hamburg. Plant Cell Physiol 54:1200–1216
Chen B, Walser JC, Rodgers TH, Sobota RS, Burke MK, Rose MR, Feder ME (2007) Abundant, diverse, and consequential P elements segregate in promoters of small heat-shock genes in Drosophila populations. J Evol Biol 20:2056–2066
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
da Hao C, Ge G, Xiao P, Zhang Y, Yang L (2011) The first insight into the tissue specific taxus transcriptome via Illumina second generation sequencing. PLoS One 6:e21220
Fullwood MJ, Wei CL, Liu ET, Ruan Y (2009) Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 19:521–532
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Gracey AY, Fraser EJ, Li W, Fang Y, Taylor RR, Rogers J, Brass A, Cossins AR (2004) Coping with cold: an integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc Natl Acad Sci USA 101:16970–16975
Gulick PJ, Drouin S, Yu Z, Danyluk J, Poisson G, Monroy AF, Sarhan F (2005) Transcriptome comparison of winter and spring wheat responding to low temperature. Genome 48:913–923
Guy CL, Li QB (1998) The organization and evolution of the spinach stress 70 molecular chaperone gene family. Plant Cell 10:539–556
He CY, Cui K, Zhang JG, Duan AG, Zeng YF (2013) Next-generation sequencing-based mRNA and microRNA expression profiling analysis revealed pathways involved in the rapid growth of developing culms in Moso bamboo. BMC Plant Biol 13:119
Hiz MC, Canher B, Niron H, Turet M (2014) Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One 9:e92598
Hsieh EJ, Cheng MC, Lin TP (2013) Functional characterization of an abiotic stress-inducible transcription factor AtERF53 in Arabidopsis thaliana. Plant Mol Biol 82:223–237
Hu J, You F, Wang Q, Weng S, Liu H, Wang L, Zhang PJ, Tan X (2014) Transcriptional responses of olive flounder (Paralichthys olivaceus) to low temperature. PLoS One 9:e108582
Kang L, Chen B, Wei JN, Liu TX (2009) Roles of thermal adaptation and chemical ecology in Liriomyza distribution and control. Annu Rev Entomol 54:127–145
Li QB, Haskell DW, Guy CL (1999) Coordinate and non-coordinate expression of the stress 70 family and other molecular chaperones at high and low temperature in spinach and tomato. Plant Mol Biol 39:21–34
Lin BL, Wang JS, Liu HC, Chen RW, Meyer Y, Barakat A, Delseny M (2001) Genomic analysis of the Hsp70 superfamily in Arabidopsis thaliana. Cell Stress Chaperones 6:201–208
Liu S, Kuang H, Lai Z (2014) Transcriptome analysis by Illumina high-throughout paired-end sequencing reveals the complexity of differential gene expression during in vitro plantlet growth and flowering in Amaranthus tricolor L. PLoS One 9:e100919
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Mishkind M, Vermeer JE, Darwish E, Munnik T (2009) Heat stress activates phospholipase D and triggers PIP accumulation at the plasma membrane and nucleus. Plant J 60:10–21
Montero-Barrientos M, Hermosa R, Cardoza RE, Gutierrez S, Nicolas C, Monte E (2010) Transgenic expression of the Trichoderma harzianum hsp70 gene increases Arabidopsis resistance to heat and other abiotic stresses. J Plant Physiol 167:659–665
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Natarajan P, Parani M (2011) De novo assembly and transcriptome analysis of five major tissues of Jatropha curcas L. using GS FLX titanium platform of 454 pyrosequencing. BMC Genom 12:191
Oono Y, Yazawa T, Kawahara Y, Kanamori H, Kobayashi F, Sasaki H, Mori S, Wu J, Handa H, Itoh T, Matsumoto T (2014) Genome-wide transcriptome analysis reveals that cadmium stress signaling controls the expression of genes in drought stress signal pathways in rice. PLoS One 9:e96946
Pan J, Tristram-Nagle S, Kucerka N, Nagle JF (2008) Temperature dependence of structure, bending rigidity, and bilayer interactions of dioleoylphosphatidylcholine bilayers. Biophys J 94:117–124
Rocheta M, Becker JD, Coito JL, Carvalho L, Amancio S (2014) Heat and water stress induce unique transcriptional signatures of heat-shock proteins and transcription factors in grapevine. Funct Integr Genomics 14:135–148
Saidi Y, Finka A, Muriset M, Bromberg Z, Weiss YG, Maathuis FJ, Goloubinoff P (2009) The heat shock response in moss plants is regulated by specific calcium-permeable channels in the plasma membrane. Plant Cell 21:2829–2843
Scott GR, Johnston IA (2012) Temperature during embryonic development has persistent effects on thermal acclimation capacity in zebrafish. Proc Natl Acad Sci USA 109:14247–14252
Simon SA, Advani S, McIntosh TJ (1995) Temperature dependence of the repulsive pressure between phosphatidylcholine bilayers. Biophys J 69:1473–1483
Sokabe T, Tsujiuchi S, Kadowaki T, Tominaga M (2008) Drosophila painless is a Ca2+-requiring channel activated by noxious heat. J Neurosci 28:9929–9938
Somero GN (1995) Proteins and temperature. Annu Rev Physiol 57:43–68
Song X, Ge T, Li Y, Hou X (2015) Genome-wide identification of SSR and SNP markers from the non-heading Chinese cabbage for comparative genomic analyses. BMC Genom 16:328
Sung DY, Vierling E, Guy CL (2001) Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiol 126:789–800
Swindell WR, Huebner M, Weber AP (2007) Plastic and adaptive gene expression patterns associated with temperature stress in Arabidopsis thaliana. Heredity (Edinb) 99:143–150
Ueda MJ, Ito T, Okada TS, Ohnishi SI (1976) A correlation between membrane fluidity and the critical temperature for cell adhesion. J Cell Biol 71:670–674
Van Verk MC, Hickman R, Pieterse CM, Van Wees SC (2013) RNA-Seq: revelation of the messengers. Trends Plant Sci 18:175–179
Walser JC, Chen B, Feder ME (2006) Heat-shock promoters: targets for evolution by P transposable elements in Drosophila. PLoS Genet 2:e165
Wei W, Qi X, Wang L, Zhang Y, Hua W, Li D, Lv H, Zhang X (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom 12:451
Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8:206–216
Xiao B, Coste B, Mathur J, Patapoutian A (2011) Temperature-dependent STIM1 activation induces Ca2+ influx and modulates gene expression. Nat Chem Biol 7:351–358
Xiong Y, Li QB, Kang BH, Chourey P (2011) Discovery of genes expressed in basal endosperm transfer cells in maize using 454 transcriptome sequencing. Plant Mol Biol Rep 29:835–847
Xu J, Xue C, Xue D, Zhao J, Gai J, Guo N, Xing H (2013) Overexpression of GmHsp90 s, a heat shock protein 90 (Hsp90) gene family cloning from soybean, decrease damage of abiotic stresses in Arabidopsis thaliana. PLoS One 8:e69810
Yang N, Yue XL, Chen XL, Wu GF, Zhang TG, An LZ (2012) Molecular cloning and partial characterization of a novel phospholipase D gene from Chorispora bungeana. Plant Cell Tissue Organ Culture (PCTOC) 108:201–212
Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, Zhuang R, Lu Z, He Z, Fang X, Chen L, Tian W, Tao Y, Kristiansen K, Zhang X, Li S, Yang H, Wang J, Wang J (2010) Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20:646–654
Zhang X, Li J, Liu A, Zou J, Zhou X, Xiang J, Rerksiri W, Peng Y, Xiong X, Chen X (2012) Expression profile in rice panicle: insights into heat response mechanism at reproductive stage. PLoS One 7:e49652
Acknowledgments
This study was funded by the National Key Science and Technology Supporting Program of China (Grant No. 2013BAD01B04-11) and by the Jiangsu agricultural science and technology innovation fund of China [Grant No. CX (13) 2004]. We also thank Genepioneer Biotechnologies for their help with the bioinformatics analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Not relevant.
Additional information
Communicated by W. Zhou.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, H., Chen, L., Song, B. et al. De novo transcriptome sequencing of pakchoi (Brassica rapa L. chinensis) reveals the key genes related to the response of heat stress. Acta Physiol Plant 38, 252 (2016). https://doi.org/10.1007/s11738-016-2269-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-016-2269-5