
Abstract
Traditionally, simple sequence repeat (SSR) markers have been developed from libraries of genomic DNA. However, the large, repetitive nature of conifer genomes makes development of robust, single-copy SSR markers from genomic DNA difficult. Expressed sequence tags (ESTs), or sequences of messenger RNA, offer the opportunity to exploit single, low-copy, conserved sequence motifs for SSR development. From a 20,275-unigene spruce EST set, we identified 44 candidate EST-SSR markers. Of these, 25 amplified and were polymorphic in white, Sitka, and black spruce; 20 amplified in all 23 spruce species tested; the remaining five amplified in all except one species. In addition, 101 previously described spruce SSRs (mostly developed from genomic DNA), were tested. Of these, 17 amplified across white, Sitka, and black spruce. The 25 EST-SSRs had approximately 9% less heterozygosity than the 17 genomic-derived SSRs (mean H=0.65 vs 0.72), but appeared to have less null alleles, as evidenced by much lower apparent inbreeding (mean F=0.046 vs 0.126). These robust SSRs are of particular use in comparative studies, and as the EST-SSRs are within the expressed portion of the genome, they are more likely to be associated with a particular gene of interest, improving their utility for quantitative trait loci mapping and allowing detection of selective sweeps at specific genes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Molecular genetic markers are variable regions of DNA that provide valuable genetic tools in genetic linkage mapping, association studies, phylogeographic studies, and for the estimation of several population genetic parameters, such as diversity, gene flow, and inbreeding (Bruford and Wayne 1993). To date, the molecular markers most widely applied to tree species have been isozymes, random fragment length polymorphisms (RFLPs), random amplified polymorphic DNA (RAPDs), amplified fragment length polymorphisms (AFLPs), and simple sequence repeats (SSRs). Each marker technique has attributes that offer different advantages (Ritland and Ritland 2000). Isozymes are well studied and established, but are not numerous or highly polymorphic. RFLPs utilize probes derived from either genomic or coding DNA (cDNA) and are codominant markers, but require a large amount of high quality DNA. RAPDs and AFLPs do not require any sequence knowledge of the genome, and so are easy to apply to uncharacterized genomes. However, they are usually dominant and are often difficult to transfer between different mapping populations or species. Additionally, RAPDs are notoriously difficult to transfer across laboratories (Jones et al. 1997).
SSR markers exhibit codominance and are usually highly polymorphic, and thus, seem to be the ideal marker (Ritland and Ritland 2000). However, their development requires a significant investment, and their cross-species transferability is normally quite limited due to either disappearance of the repeat region, or to degeneration of the primer binding sites. Traditionally, the first stage of SSR marker development is to probe a genomic library with a particular SSR oligonucleotide and sequence positive clones. However, the success rate of identifying robust SSR markers from genomic DNA is typically low due to the high proportion of primers that do not amplify genetically interpretable PCR fragments (Squirrell et al. 2003).
In conifers, SSR discovery from genomic libraries (van de Ven and McNicol 1996; Pfeiffer et al. 1997; Rajora et al. 2000; Hodgetts et al. 2001; Scotti et al. 2002a, b) has been a particularly difficult process, with very low success rate, probably because of the large, repetitive nature of their genomes (Pfeiffer et al. 1997; Bérubé et al. 2003). Despite these problems, SSRs remain the marker system of choice for a number of conifer mapping projects (Paglia and Morgante 1998).
Expressed sequence tags (ESTs) are sequenced portions of messenger RNA and offer an alternative route for SSR marker discovery, particularly for the repetitive genomes found in conifers. The advent of large-scale databases with tens of thousands of ESTs provides resources for the novel, “in silico” identification of genetic markers. In marker development, EST databases have largely been used for identification of single nucleotide polymorphisms (SNPs) (Rafalski 2002). However, SSRs are found in both the untranslated regions of ESTs and occasionally within coding regions (Cardle et al. 2000).
One advantage of these “EST-SSRs” is that they are directly associated with a coding gene, and so may be useful for association with phenotypic traits. Also, because EST sequences are evolutionary conserved, cross-species PCR amplification of EST-SSRs are expected to be more successful compared to SSRs developed from genomic DNA (Arnold et al. 2002; Saha et al. 2003); however, their levels of variability may not be as great due to selective constraints. Finally, with their relatively high levels of variability, EST-SSRs seem especially appropriate for the detection of selective sweeps (Vigouroux et al. 2002).
Here, we utilize an EST database—developed as part of the Genome British Columbia Forestry Genomics project—to identify and characterize SSR markers for spruce. This database provides a valuable and unique resource for the development of new SSR markers within spruce and also for comparative analysis of genome structure and organization. We report 25 new EST–SSR markers of primary use with white, Sitka, and black spruce. We also evaluate 101 previously reported spruce SSRs (derived from genomic DNA libraries), evaluate the use of all SSRs across 23 spruce species, and arrive at a total set of 42 robust microsatellites markers for spruce.
Materials and methods
Library construction and DNA sequencing
Nine directional cDNA libraries were constructed from a range of tissues (xylem, phloem, bark, foliage, and roots) at different developmental stages of seedlings and mature trees, as well as from trees or seedlings exposed to chemical elicitors (methyl jasmonate), or mechanical wounding. Tissues were obtained from three different spruce species: white spruce (Picea glauca) cultivar PG29, Sitka spruce (Picea sitchensis) cultivar Gb2-229, and the interior spruce (P. glauca × Picea engelmannii) cultivar Fal-1028. cDNA libraries were constructed (5′ EcoRI, 3′ XhoI) using the pBluescript II XR cDNA Library Construction Kit, following manufacturer’s instructions with modifications (Stratagene). Select cDNA libraries were normalized according to the Soares method (Soares et al. 1994). A complete technical description of library construction methods will be reported elsewhere.
Library-stock plasmid DNA was transformed into electrocompetent DH10B T1-phage-resistant Escherichia coli cells (Invitrogen) and robotically arrayed into 384-well plates from which glycerol stocks were prepared. Plasmid DNA was extracted from overnight 96-well cultures and BigDye Terminator (ABI) cycle sequenced on an ABI Prism 3700 DNA Analyzer, using conventional procedures and the −21 M13 forward primer (5′-TGTAAAACGACGGCCAGT-3′) to obtain predominantly 3′ end sequences. DNA sequence chromatograms were processed using the PHRED software (Ewing and Green 1998; Ewing et al. 1998). Sequences were quality trimmed according to the high-quality contiguous region determined by PHRED and then vector trimmed using CROSS_MATCH software (http://www.phrap.org). Sequences with less than 70 quality bases after trimming were discarded.
EST database and SSR search
The EST database used for this search consisted of 34,846 EST sequences, which were quality clipped using PHRED and our own in-house software “EST Clean.” This step also removed poly-A tail sequence from the ESTs. The clipped sequences were aligned to generate 20,275 unigenes, using the CAP-3 software package (Huang and Madan 1999). We developed an EST–SSR discovery software package (BuildSSR, available at http://www.genetics.forestry.ubc.ca/ritland/programs.html) to search for SSRs within this unigene set. This included database organization, repeat-finding software, and tools for SSR-distribution analysis. This SSR-discovery pipeline identifies SSRs in the unigene set, constructs a summary table, and then, builds a FASTA-format database that includes the repeat type, size, and position. A minimum perfect-repeat number of nine dinucleotide repeats, six trinucleotide repeats, and four tetranucleotide repeats was used for the search. EST containing SSRs were then annotated using BLAST software.
Primer design and PCR conditions
Primers spanning 44 EST-SSRs were designed. These repeats were detected in sequences from the interior, white, and Sitka spruce libraries. Primers were designed using the Primer 3 software (Rozen and Skaletsky 2000). Regions 50 bp from each end of the repeat were excluded from primer site consideration, and all primers were designed to have similar annealing temperatures to allow for uniform PCR cycling conditions. The forward primer was tailed with an M13 sequence (Oetting et al. 1995) to facilitate visualization of PCR products on a LiCor 4200 (LiCor, Lincoln, Neb., USA).
The 101 previously described spruce SSR primers derived from the genomic DNA approach were also synthesized (Pfeiffer et al. 1997; Rajora et al. 2000; Scotti et al. 2000; Hodgetts et al. 2001; Scotti et al. 2002a, b; C. Newton, personal communication), with the forward primer tailed with an M13 sequence as above.
PCR was performed with 25 ng genomic DNA, 0.2 μM of each forward and reverse primer, 0.05 μM M13 IRD labeled primer, 0.2 mM dNTPs, 1.5 mM MgCl2, and 1 U of AmpliTaq DNA polymerase (Roche) in a 20-μl volume. PCR cycling conditions consisted of an initial denaturation step of 95°C for 2 min; 30 cycles of 95°C for 20 s, 53°C for 20 s, and 72°C for 30 s; followed by a final extension step of 72°C for 3 min.
Plant material
Fresh needle tissue from the current year’s growth was collected from 20 mature trees in wild populations of both white and Sitka spruce. The white spruce population was sampled in the region surrounding the town of Fort Nelson, situated in the northeast corner of the province of British Columbia, Canada. Trees of this population were sampled 1–2 km apart. The Sitka spruce population was located on Kodiak Island, Alaska, which marks the northern migrating tip of the species’ range. Trees of this population were sampled 30–50 m apart. DNA from 20 black spruce individuals was obtained from samples collected in Manitoba and Saskatchewan, Canada. Buds were collected from a single tree from each of 23 spruce species (for list see Table 3), which were growing as a collection at the British Columbia Ministry of Forests Kalamalka research station, Vernon, B.C. Only one genetic individual was available for each species in this collection. DNA was isolated from the bud and needle tissue following the CTAB method described by Doyle and Doyle (1990).
SSR testing and assay
The 44 SSR developed from the EST database were tested on the above described population collections of Sitka, white, and black spruce. In addition, the 101 previously described SSR primer pairs were tested on a panel of two white, two Sitka, one black, and one red spruce individual. The SSR primer pairs that amplified products from these species were then tested on the collection of 23 spruce species.
In the testing and assay, presence or absence of microsatellite PCR products was scored on 2% agarose gels. When products were found, they were tested for polymorphism on 6% (Long Ranger) polyacrylamide gels, using a LiCor 4200 automated sequencer. Microsatellite products were detected by M13 tailed primer (Oetting et al. 1995).
Analyses
Observed heterozygosity (Ho), expected heterozygosity (He) and the inbreeding coefficient (F: F=1−Ho/He) were estimated for each SSR locus within each of the three spruce species (Sitka, white, black). Standard errors of F were determined by bootstrapping individuals within populations, using a Fortran 95 program written by K. Ritland.
Genetic distances between individuals in the 23 species set were estimated as the mean squared difference of allele sizes (Goldstein et al. 1995)—after sizes were normalized by dividing by the variance of allele size (specific for each locus)—using a Fortran 95 program written by K. Ritland. A total of 100 bootstrap datasets were constructed by resampling loci. For each replicate, the computer program NEIGHBOR (in PHYLIP, Phylogeny Inference Package, version 3.57c, Felsenstein 1995) was used to construct an unrooted tree, using the neighbor-joining (NJ) method (Saitou and Nei 1987). The 100 trees were then evaluated by CONSENSE (in PHYLIP) to find an overall consensus tree, with confidence numbers attached to each branch.
The metric of mean squared allele size difference outperforms heterozygosity at differences over larger time periods [≥1,000 generations, particularly when standardized (Neff 2004)]. Hence, instead of standard measures such as Nei’s genetic distance or the proportion of bands not shared, we used a mean squared allele size difference, standardized by mutation rate (as the variance of allele size is proportional to the mutation rate, c.f. Goldstein et al. 1995).
Results
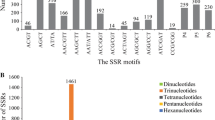
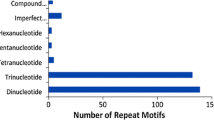
From the Genome British Columbia (BC) spruce EST unigene database, 188 unique SSR sequences were found within 183 contigs. A total of 119 dinucleotide, 61 trinucleotide, and eight tetranucleotide repeats were found (Fig. 1). The most common class of repeat was AT (91 of 188 SSRs). Of the SSR sequences found in the EST database, 31 were at the extreme 3′ end of the ESTs (adjoining the poly-A tail) and 22 were at the 5′ end of the sequences; therefore, primers could not be designed for these sequences. The distribution of the SSR repeat types in relation to the coding sequence was non-random. Of the 31 SSRs at the 3′ end of the ESTs, 30 were AT repeats (the remaining SSR was an ATT repeat). Of the 22 repeat types at the 5′ end of the ESTs, 19 were AG repeats (the remaining SSRs were one each of AT, GAC and AGA). Only two AC repeats were identified within the EST database. G+C content within the SSR containing ESTs was 40.2%, which is comparable to the G+C content in and Arabidopsis SSR containing ESTs (43.8%) (Morgante et al. 2002).

Distribution of simple sequence repeat motifs in the Genome British Columbia Forestry spruce EST database
Of the 145 primer pairs, 41 detected a single locus, and one previously developed SSR detected two loci across these four species (Table 1). This set of 42 primer pairs included 25 EST-SSRs and 17 previously developed SSRs.
HeHo heterozygosity, and F are shown in Table 2 (in some cases there were insufficient individuals to obtain adequate estimates of F). As is normal with microsatellites, heterozygosity varied widely among loci. The average heterozygosity was highest in white spruce (0.78), lower in black spruce (0.72), and lowest in Sitka spruce (0.55).
The EST-SSRs showed significantly less variation than the genomic-derived SSRs; He values were 6.25% less in white spruce, 15% less in black spruce, and 9% less in Sitka spruce. Likewise, the numbers of alleles at ESR-SSR loci were comparably lower in all three species. Interestingly, F values were significantly lower at the EST-SSR compared to the genomic-derived loci (0.02 vs 0.13 in Sitka, 0.03 vs 0.10 in white, and 0.09 vs 0.14 in black).
Of the 43 loci amplified by the 42 primer pairs determined to be informative across white, Sitka, black, and red spruce, 33 were identified in all 23 spruce species (Fig. 2; Table 3). The minimum number of species in which a particular locus was present was 17. Twenty-five EST–SSRs primer pairs developed from the Genome BC spruce EST database were included in this set of markers. Of these, 20 amplified single locus markers from across all 23 spruce species tested, while five amplified single-locus markers from 22 of the 23 spruce species tested.

Amplification of locus WS0092.A19 across the 23 spruce species
Figure 3 gives the NJ tree of microsatellite genetic distances among the 23 spruce species, and Fig. 4 gives the consensus tree of microsatellite genetic distances among 23 spruce species. The relatively deep rooting of each species is due to the variability and high evolutionary rate at SSR loci. While some clustering of related species is evident, bootstrap confidence levels are not high.

Neighbor-joining tree of microsatellite genetic distances among the 23 spruce species

Consensus tree of microsatellite genetic distances among 23 spruce species. The numbers at the forks indicate the number of times the group consisting of the species, which are to the right of that fork occurred among the trees, out of 100 trees
Discussion
The EST–SSR markers are adjacent to coding genes, and the function of these genes can be often identified via sequence similarity to annotated genes in other plant species. Thus, they are useful in quantitative trait locus mapping and particularly “genomic scans” (Vigouroux et al. 2002). Their association with coding genes makes EST-SSRs more likely to be single copy, which is particularly useful for species with large genomes such as spruce. Furthermore, as coding regions tend to be more conserved, this potentially increases the transferability of these EST-SSRs across spruce species. While the EST-derived SSR markers in this study were somewhat less variable than the genomic SSR markers, the F values were also significantly lower, suggesting a lower frequency of troublesome null alleles in EST-SSRs.
SSR locations in spruce ESTs
The SSRs exhibited differential distribution within the expressed sequences. AT repeats were preferentially found at the 3′ end of the EST sequences, while AG repeats were preferentially found at the 5′ end of sequences. While Scotti et al. (2000) found six AC repeat regions from a Norway spruce cDNA library clustered at the 3′ end of the expressed sequences, we found only two AC repeats within our 3′ EST collection. This may reflect a difference in SSR composition between Norway spruce and the North American spruces used for our cDNA libraries. Alternatively, by specifically targeting AC repeats, Scotti et al. (2000) may have identified the rare AC repeats found in expressed portions of the spruce genome.
SSR motif types in spruce ESTs
The most common SSR motif found in our EST database was AT, accounting for 91 of the 188 repeats identified. By contrast, in Arabidopsis ESTs, AAG is the most common class of SSR, and AT repeats are less prevalent (Cardle et al. 2000). AT repeats, however, are the second most common SSR type (after poly A repeats) in Arabidopsis and other plant genomic DNA (Cardle et al. 2000). The prevalence of AT repeats in spruce ESTs is also supported by SSR searches of Picea sequences in the EMBL database, where of seven SSRs developed, four were AT repeats (Besnard et al. 2003). This prevalence of AT repeats in spruce ESTs may be a hitherto unnoted feature, as other studies where SSRs have been isolated from spruce coding sequences have utilized specific repeat probes (not AT) (e.g., Scotti et al. 2000). Alternatively, as AT repeats were found to be preferentially clustered at the 3′ end of the ESTs, this preponderance of AT repeats may be a consequence of the 3′ sequencing of this EST database. We are currently in the process of obtaining full-length EST sequences, and a survey of these may reveal a different distribution of SSR repeats.
SSR polymorphism
The amplification of the SSRs in Sitka, white, and black spruce populations revealed high levels of polymorphism, as indicated by the high average number of alleles and the high He and Ho, both typical of SSR markers. This suggests that most of these SSR markers will be useful in parentage and clonal assessments because of their high potential for discrimination. They will also be useful in constructing genetic linkage maps, as these markers will likely be segregating in a range of crosses.
The F for specific SSRs allows identification of loci with putative null alleles, with those showing significantly higher F values indicating the presence of null alleles. Null alleles can bias estimates of genetic variation and genetic structure, and are not useful for genetic mapping. Also, loci with prominent stutter bands often exhibited higher F values, due to the difficulty in scoring of heterozygous genotypes for adjacent sized alleles. In contrast to the genomic DNA-derived markers, our EST–SSR markers gave more uniform F values the three spruce species. Two genomic-derived loci in particular showed consistent patterns across the three species: PAAC 19 (positive F) and UAPgAG150A (negative F).
Cross-species amplification of SSR markers
Of the 43 loci identified as informative in white, Sitka, black, and red spruce, the majority (33/43) were able to amplify alleles across all 23 spruce species tested. The minimum number of species in which a particular locus was identified was 17 (locus SPL3AG1A4). This suggests that the regions flanking the SSRs are well conserved across the spruce species tested, and that if a particular locus can be amplified from white, Sitka, black, and red spruce, then it is likely that the locus will be widely transferable throughout other spruce species as well. Of the 44 SSR markers developed from the EST database, 25 were informative in white, Sitka, and black spruce. From these 25 loci, 20 were identified in all 23 spruce species tested, while the remaining five loci were detected in 22 of the 23 species.
While SSRs are instrumental in genetic mapping (e.g., Dib et al. 1996), studies of kinship (e.g., Queller et al. 1993), and population structure (e.g., Bowcock et al. 1994), they have received limited use as a tool for phylogenetic reconstruction of closely related species (reviewed by Schlötterer 2001). This is mainly due to allele size homoplasy resulting from an exceptionally high mutation rate. However, when a genetic distance measure that takes into account the mutational process is used (Goldstein et al. 1995; Neff 2004), SSRs, particularly those developed from ESTs, can be potentially very informative in resolving newly diverged specific complexes or groups with slower rates of evolution.
Interestingly, the tree topology obtained by microsatellite genetic distances among species (Figs. 3, 4) was similar to that obtained from phenetic and cladistic analyses of chloroplast DNA RFLPs (Sigurgeirsson and Szmidt 1993). Highlights of this similarity include P. mexicana and P. glauca clustering together in congruence with Sigurgeirsson and Szmidt’s (1993) “P. glauca alliance” and the association of P. asperta, P. koyamai, and P. koraiensis. Results from the bootstrap routine, however, showed no support for the branches of the phylogenetic tree generated. This is most likely a product of sampling a single individual per tree species. Because portions of the phylogenetic tree obtained matched the results of Sigurgeirsson and Szmidt (1993) and was in agreement with generally accepted views of classification within Picea, we propose that the microsatellite markers tested in this study, if applied to multiple individuals of each species, will likely prove to be powerful tools for investigating phylogenetic relationships within Picea.
Comparison of EST-derived SSRs with other SSRs in spruce
In this study, we found that the use of an EST database to develop novel SSR markers led to a high rate of success when compared to other studies. In addition, the EST–SSR markers developed and presented here have been readily transferable across species. Of 44 EST-SSRs, 25 were widely transferable across spruce species (~57%), while only 17 of 101 previously developed SSR markers were as widely transferable (~17%). These SSR markers are in the process of being placed onto a genetic linkage map of white spruce. This will increase their usefulness for other purposes such as population studies because markers evenly spaced throughout the genome will be able to be chosen. Also, the large allele size difference between different loci will allow placement of loci into “bins” for multiplexing. Although the potential for coamplification of loci has not been tested yet, even post-PCR pooling of these loci will save time and money by reducing the number of gels that have to be run.
Previous attempts at developing SSR markers from conifer genomic sequences have been hampered by a low success rate due to many primer pairs yielding complex banding patterns that cannot be genetically interpreted. This has been attributed to the large proportion of repetitive or low complexity sequence present in conifer genomes (Pfeiffer et al. 1997). The use of an EST database to identify SSR markers has resulted in the development of a higher proportion of useful and informative loci. This study identified a preponderance of AT repeats from spruce ESTs, in contrast to other plant genomes. When a full-length EST database is available for spruce, we will be able to determine if this SSR distribution is confirmed or if it is an artefact caused by the 3′-sequence data currently in our EST database.
References
Arnold C, Rossetto M, McNally J, Henry RJ (2002) The application of SSRs characterized for grape (Vitis vinifera) to conservation studies in Vitaceae. Am J Bot 89:22–28
Bérubé Y, Ritland CE, Ritland K (2003) Isolation, characterization, and cross-species utility of microsatellites in yellow cedar (Chamaecyparis nootkatensis). Genome 46:353–361
Besnard G, Achere V, Faivre Rampant P, Favre JM, Jeandroz S (2003) A set of cross-species amplifying microsatellite markers developed from DNA sequence databanks in Picea (Pinaceae). Mol Ecol Notes 3:380–383
Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL (1994) High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457
Bruford MW, Wayne RK (1993) Microsatellites and their application to population genetic studies. Curr Opin Genet Dev 3:939–943
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 23:13–15
Ewing B, Green P (1998) Base-calling of automated sequencer traces using PHRED II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using PHRED. I. Accuracy assessment. Genome Res 8:175–185
Felsenstein J (1995) PHYLIP (Phylogeny Inference Package), ver 3.57c, University of Washington
Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–471
Hodgetts RB, Aleksiuk MA, Brown A, Clarke C, Macdonald E, Nadeem S, Khasa D (2001) Development of microsatellite markers for white spruce (Picea glauca) and related species. Theor Appl Genet 102:1252–1258
Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9:868–877
Jones CJ, Edwards KJ, Castaglione S, Winfield MO, Sala F, van de Wiel C, Bredemeijer G, Vosman B, Matthes M, Daly A, Brettschneider R, Bettini P, Buiatti M, Maestri E, Malcevschi A, Marmiroli N, Aert R, Volckaert G, Rueda J, Linacero R, Vazquez A, Karp A (1997) Reproducibility testing of RAPD, AFLP and SSR markers in plants by a network of European laboratories. Mol Breed 3:381–390
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Neff BD (2004) Mean d2 and divergence time: transformations and standardizations. J Hered 95:165–171
Oetting WS, Lee HK, Flanders DJ, Wiesner GL, Sellers TA, King RA (1995) Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics 30:450–458
Paglia G, Morgante M (1998) PCR-based multiplex DNA fingerprinting techniques for the analysis of conifer genomes. Mol Breed 4:173–177
Pfeiffer A, Olivieri AM, Morgante M (1997) Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 40:411–419
Queller DC, Strassmann JE, Hughes CR (1993) Microsatellites and kinship. Trends Ecol Evol 8:285–288
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5:94–100
Rajora OP, Rahman MH, Dayanandan S, Mosseler A (2000) Isolation, characterization, inheritance and linkage of microsatellite DNA markers in white spruce (Picea glauca) and their usefulness in other spruce species. Mol Gen Genet 264:871–882
Ritland C, Ritland K (2000) DNA fragment markers in plants. In: Baker AJ (ed) Molecular methods in ecology. Blackwell, Oxford, pp 208–234
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Saha S, Karaca M, Jenkins JN, Zipf AE, Reddy OUK, Kantety RV (2003) Simple sequence repeats as useful resources to study transcribed genes of cotton. Euphytica 130:355–364
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schlötterer C (2001) Genealogical inference of closely related species based on microsatellites. Genet Res 78:209–212
Scotti I, Magni F, Fink R, Powell W, Binelli G, Hedley PE (2000) Microsatellite repeats are not randomly distributed within Norway spruce (Picea abies K.) expressed sequences. Genome 43:41–46
Scotti I, Magni F, Paglia G, Morgante M (2002a) Trinucleotide microsatellites in Norway spruce (Picea abies): their features and the development of molecular markers. Theor Appl Genet 106:40–50
Scotti I, Paglia G, Magni F, Morgante M (2002b) Efficient development of dinucleotide microsatellite markers in Norway spruce (Picea abies Karst.) through dot-blot selection. Theor Appl Genet 104:1035–1041
Sigurgeirsson A, Szmidt AE (1993) Phylogenetic and biogeographic implications of chloroplast DNA variation in Picea. Nord J Bot 13:233–246
Soares MB, Bonaldo MF, Jelene, P, Su L, Lawton L, Efstratiadis A (1994) Construction and characterization of a normalized cDNA library. Proc Natl Acad Sci USA 91:9228–9232
Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, Powell W (2003) How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol 12:1339–1348
Ven WTG van de, McNicol RJ (1996) Microsatellites as DNA markers in Sitka spruce. Theor Appl Genet 93:613–617
Vigouroux Y, McMullen M, Hittinger CT, Houchins K, Schulz L, Kresovich S, Matsuoka Y, Doebley J (2002) Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication Proc Natl Acad Sci USA 99:9650–9655
Acknowledgements
Genome Canada and the Province of British Columbia, through the Genome BC Forestry Genome Project, funded this research. We acknowledge the support of the Vancouver Genome Sciences Centre for EST sequencing and database development. We thank Dr. Sally Aitken (University of British Columbia) for the white spruce collections, Washington Gapare (University of British Columbia) for the Sitka spruce collections, and Dr. Om Rajora (Dalhousie University) for the black spruce collections. Dr. Barry Jaquish of the B.C. Ministry of Forests Kalamalka research station provided the spruce species collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by O. Savolainen
Rights and permissions
About this article
Cite this article
Rungis, D., Bérubé, Y., Zhang, J. et al. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor Appl Genet 109, 1283–1294 (2004). https://doi.org/10.1007/s00122-004-1742-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-004-1742-5