
Abstract
Ear row number (ERN) is not only a key trait involved in maize (Zea mays L.) evolution but also an important component directly related to grain yield. In this report, 325 recombinant inbred lines (RILs, F6:7) derived from a cross between B73 with 16 rows and SICAU1212 with four rows (two-ranked with two rows per rank) were utilized to detect quantitative trait loci (QTL) associated with ERN and two-ranked versus many-ranked ears (TR). Compared to modern maize that formed approximately 8–20 rows, SICAU1212 with four rows was the extreme case. A total of 12 and 8 QTLs were associated with ERN and TR across four environments through single-environment mapping, respectively. Each QTL responsible for ERN explained 2.33–21.28 % of the phenotypic variation. And the TR variation contributed by individual TR QTL ranged from 2.09 to 12.99 %. Notably, only three QTLs, qERN2-1 (bin 2.02), qERN8-1 (bin 8.02) and qERN8-2 (bin 8.04), were consistently detected in each environment and by joint analysis among all environments, which simultaneously influenced ERN and TR. One of the three QTLs, qERN8-1 was also identified as interacting with environment. In addition, nine pairs of significant epistatic interactions (two for ERN and seven for TR) were detected among all QTLs. The epistasis between qTR2-1 and qTR8-1 was consistent in most environments. This present study may provide the understanding of the genetic basis of ERN and TR and a foundation for further fine-mapping of these common QTLs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The growing population exerts great pressure on the achievement of further improvement in grain yield. Compared with other crops, maize (Zea mays L.) presents better ideal plant type in terms of the structure of the photosynthetic reaction and it plays a crucial role in securing the world’s food supply. Therefore, the inheritance of grain yield of maize has been the focus of many studies by agricultural scientists and plant breeders (Collard and Mackill 2008). As a complex quantitative trait, however, maize grain yield is influenced by many genetic and environmental factors due to its relatively low heritability (Beavis et al. 1994; Yan et al. 2006). As an important yield determinant, the ear row number (ERN) exhibited significantly positive correlation with the grain yield (Li et al. 2009; Lu et al. 2011; Sreckov et al. 2011). Meanwhile, the inheritance of ERN presented a significant additive effect and was less influenced by situational factors (Dofing et al. 1991; Leng 1963; Petrovic 1998). Thus, maize grain yield could be dissected into its several related components, and its genetic basis could be preliminarily explained by the effects of QTLs that control its components such as ERN.
Previously, maize × teosinte populations including F2 (Doebley 1992; Doebley and Stec 1991, 1993; Doebley et al. 1990; Edwards et al. 1987; Stuber et al. 1987), BC1 (Briggs et al. 2007), TC1 (Lauter and Doebley 2002) and nearly isogenic recombinant inbred lines (NIRILs) (Lemmon and Doebley 2014) were used for the detection of loci, which controlled the changes in ERN during early domestication. Among these studies, the most consistent result was that a certain region on the short segment of chromosome 2 of maize had a large effect on ERN. In addition, a continuous distribution of two-ranked versus many-ranked ears (TR) value was observed in segregating populations derived from maize to teosinte hybrids. TR was considered to be one of the key morphological traits that distinguish maize and teosinte ears (Doebley 2004). Langham (1940) firstly reported tr1 gene controlling TR, which had a large effect on the TR (Doebley and Stec 1991). Nevertheless, several inconsistent results have been pointed out, namely regions of chromosomes 3, 5 and 6 also had strong effects on this trait (Rogers 1950; Szabó and Burr 1996). These results suggest that the accurate location of QTL for TR could be influenced by different measurements of TR, the lack of the limited number of linkage markers and the utilization of teosintes with different genetic background.
For decades, with the rapid development of molecular genetic marker technology and quantitative genetics, diverse maize populations have been used to detect QTLs for ERN, which contribute to maize diversification, such as F2 (Yu et al. 2014), F2:3 (Cai et al. 2014; Choe and Rocheford 2012; Karen Sabadin et al. 2008; Lu et al. 2011; Veldboom and Lee 1994; Yan et al. 2006), F2:4 (Beavis et al. 1994), BC1S1 (Upadyayula et al. 2006), BC2F2 (Li et al. 2007, 2011), BC3F2:3, BC5F2:3 (Tian et al. 2014), IF2 (Tang et al. 2010), CSSLs (Li et al. 2014a) and RILs (Austin and Lee 1996; Guo et al. 2008; Liu et al. 2010). To date, few consistent QTL having large effects on ERN have been detected across diverse populations or environments. One QTL explaining 41 % of ERN variation was mapped on chromosome 2 near marker umc78 in a F2:3 lines, which was derived from a single cross between Mo17 and H99 (Veldboom and Lee 1994). More recently, a total of 36 QTLs associated with ERN and 261 significant SNPs were observed by joint linkage and genome-wide association studies (GWAS) in a nest association mapping (NAM) population, consisting of 5000 RILs from 25 families that represented the global diversity of maize (Brown et al. 2011). While numerous QTLs controlling ERN have been identified on the whole genome of maize, only three or four genes have been determined through the use of maize mutants. The use of mutator (Mu) transposon insertions in the maize gene has suggested that the activity of zfl2 may influence the ERN (Bomblies and Doebley 2006; Bomblies et al. 2003). A leucine-rich repeat receptor-like protein denoted fea2 could increase the inflorescence meristems size (Taguchi-Shiobara et al. 2001) and enhance ERN of ~3 rows (Bommert et al. 2013). Maize SBP-box transcription factors genes ub2 and ub3 affected the ERN by regulating the rate of lateral primordia initiation (Chuck et al. 2014). The molecular mechanism underlying ERN variation has only been preliminarily determined, although its genetic basis has been well understood.
In this study, a RIL segregation population derived from B73 × SICAU1212 was made to identify consistent QTL responsible for ERN and TR across different environments, detect the epistatic interaction for these traits and attempt to investigate the correlation between ERN and TR. This study could provide interesting information about the genetics underlying these traits.
Materials and methods
Plant materials and field experiments
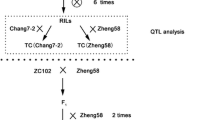
A population of 325 F6:7 RIL families was derived from a cross between inbred B73 and SICAU1212 (Fig. 1). SICAU1212 (two-ranked with two rows per rank) was derived from four-rowed waxy maize, silunuo, which was planted in Xishuangbanna, Yunnan Province of China, and subsequently self-pollinated for seven generations. Notably, silunuo with many primordial traits was considered as the progenitor of Chinese waxy corn (Tian et al. 2009).

Mature ear phenotypes illustration of B73 (left) and SICAU1212 plants (right), normal B73 ear (female inflorescence) with approximately 16 regular rows, a non-waxy and yellow kernel (endosperm) and a red cob (pericarp). The SICAU1212 plant often produces approximately four female inflorescences with approximately four rows, a waxy and white kernels and a white cob. In addition, the ear of SICAU1212 exhibits a barren patch devoid of kernels in the adaxial side of the ear. Scale bar 2 cm. (Color figure online)
The 325 RIL families used the single-seed descent and both parents were planted in a completely random design with two replications in three experimental stations located in Yunnan, Sichuan and Neimenggu Provinces of China, during 2012, 2013 and 2014, respectively. The abbreviations 12YN, 13YN, 14SC and 14NMG represented the following environments: Yunnan in 2012, Yunnan in 2013, Sichuan in 2014 and Neimenggu in 2014, respectively. Each RIL was grown in a single-row. Each row (approximately 14 plants) was 3.5 m in length with a space of 0.75 m between rows. Standard cultivation management practices and a density of 52,500 plants per ha were performed in each environment. After maturity, the ERN value in the middle point of ear was measured in individual ears (Taguchi-Shiobara et al. 2001). The ear of B73 exhibited many ranks of paired spikelets, whereas that of SICAU1212 had two ranks of paired spikelets. Some RIL ears showed an intermediate phenotype (Fig. 2). The value of the TR (%) was calculated from the following formula:

Ears exhibit the two-ranked versus many-ranked (TR). From left to right, the numbers 4/6, 4/8, 4/10, 4/12, 4/14 indicate that the TR value are 65.5, 52.8, 42.0, 44.2, 50.0, 37.5 and 48.3 %, respectively; The transition points are indicated by arrows. (Color figure online)
here, FEL is the length of ear with four rows, and EL is the whole ear length. The average ERN and TR value of ten plants from the middle of each row was utilized as the preliminary data in the analyses.
Phenotypic data analysis
Both the analyses of variance (ANOVA) and the Pearson phenotypic correlation coefficient between ERN and TR in each environment were performed using the SPSS19.0 software (http://www.spss.com). The variance components including genotype, environment, replication and interaction between genotype and environment of each trait were estimated by mixed model program. The broad-sense heritability (H 2) for each trait was estimated: H 2 = σ 2 g /(σ 2 g + σ 2 ge + σ 2 ε /rn), where σ 2 g is the genetic variance, σ 2 ge is the interaction of the genotype with the environment, σ 2 ε is the error variance, r is the number of replications, and n is the number of environments (Hallauer and Miranda 1988). As described by Knapp et al. (1985), the 90 % confidence intervals (CI) for H 2 were calculated.
Genetic linkage maps
Genomic DNA was extracted and purified using the modified CTAB procedure from the fresh leaf tissue of the parental lines and 325 RIL families’ plants (Saghai-Maroof et al. 1984). A total of 910 SSR probes chosen from the maize genome database (http://www.maizegdb.org/) were used for screening the parental lines. Of which, 107 SSR markers were finally employed to genotype the 325 RIL families. Meanwhile, 334 insertion/deletion polymorphism (Indel) markers developed by our laboratory were used to cover more of the bins within the maize genome. One hundred sixty-six of the Indel markers were added to the identification of genotype of the entire RIL families (Supplementary Fig. S1; Table S1). The 325 RIL families were eventually genotyped by 273 markers consisting of 107 SSRs markers and 166 Indels. All PCR products were eletrophoresed on 6 % denaturing polyacrylamide gels and stained with approximately 0.33 % silver nitrate (Santos et al. 1993). The linkage map was developed using Mapmaker/EXP (version 3.0b) with a linkage group at a minimal logarithm of odds (LOD) threshold of 3.00 and a maximum distance between two loci of 50 cM (Lincoln 1992). The result showed that the total length of the molecular linkage map was 1395.4 cM across the maize genome with average intervals between adjacent markers of 5.11 cM. The genomic analysis revealed that the RIL was approximately 96.05 % homozygous and 50.24 % of B73-allele. In the genetic linkage map, the orders of most molecular markers were consistent with those of their physical position. However, two exceptions were observed in this study: the Indel marker chr7-10777 was located at bin 3.07 rather than at bin 7.01 and chr4-84108 was located in bin 9.04 rather than in bin 4.05.
QTL identification
The identification of QTL for each trait in single environment was performed by the QTL IciMapping software version 3.0 (Li et al. 2008). The additive and dominant effects (ICIM-ADD) mapping method in ICIM was employed to identify QTL through stepwise regression with 1000 permutations and a walk speed of 2 cM. The CI of the QTL were estimated: CI = 163/N × R 2. Here, N is the population size, and R 2 is the value of the phenotypic variation contributed by the QTL (Darvasi and Soller 1997; Qi et al. 2011). If the LOD confidence interval of two QTLs overlapped, the corresponding QTLs were assumed to be the same one in this study. Additionally, the joint QTL, the epistatic QTL in a single environment and QTL environment interaction based on the multi-environment phenotypic values were identified by the QTL Network software version 2.0 with mixed liner model based on composite interval mapping approach (MCIM) (Yang et al. 2007). Testing window, walk speed and filtration window of genome scan were set at 10, 2 and 10 cM, respectively. The LOD threshold scores for significant QTL were obtained with a permutation test of 1000 cycles (Churchill and Doerge 1994). The letter ‘J’ was labeled into the middle of this QTL name while one QTL was only detected in joint analysis with MCIM but not in single environment through ICIM-ADD program. In the present report, if QTLs for different traits were detected within the same marker interval or their confidence intervals overlapped, the corresponding loci were assumed to be the one common QTL or QTL with pleiotropic effects.
Results
Phenotypic analysis
Compared with the ERN of B73 (16.31–17.80 rows), that of SICAU1212 presented lower value (four rows). In contrast with the ERN value of SICAU1212, its TR value (92.8–100.0 %) was always higher than that of B73 (0.0 %). In addition, B73 and SICAU1212 exhibited highly significant differences in ERN and TR, regardless of the environment (Table 1). Among the RIL families, the distributions of the numbers of ERN were approximately normal, and there were wide variations in four environments (Fig. 3), which was noteworthy that the ERN values basically presented obvious unidirectional transgressive segregation in each environment, indicating polygenic quantitative genetic control. A negative exponential distribution of TR was found in each environment (Fig. 3), seemed to indicate a potential for several major genes or oligogene influencing this phenotype (Zhou et al. 2014). The genotypic variance components (σ 2 g ) were highly significant for ERN and TR based on the ANOVA analysis, indicating the existences of significant genetic variability in RILs (Table 2). The variance of the genotype × environment interactions (σ 2 ge ), the environmental variance and the variance of replications (environment) were highly significant. The broad-sense heritabilities (H 2) of ERN and TR were estimated to be 89.46 and 92.92 %, respectively. The relatively high heritability indicated that genetic factors controlled much of the phenotypic variance. The analyses of Pearson correlation coefficient showed that the ERN significantly correlated with each other (r = 0.781–0.931) measured in different environments, as well as TR (r = 0.847–0.928) (Table 3). Notably, highly negative phenotypic correlations between ERN and TR were observed in four environments (r = −0.596 to −0.799), suggesting that there was a potential for simultaneous improvements in ERN and TR.

Distributions of the two traits value in RIL derived from B73-SICAU1212hybrids among four environments. The trait values for B73 and SICAU1212 were indicated by arrows. 12YN, 13YN, 14SC and 14NMG represented Yunnan in 2012, Yunnan in 2013, Sichuan in 2014 and Neimenggu in 2014, respectively. (Color figure online)
Identification of QTLs for ERN and TR
A total of 12 QTLs located on chromosomes 1, 2, 4, 5, 8 and 10 were associated with ERN through single-environment mapping (Table 4; Fig. 4). Each QTL accounted for 2.33–21.28 % of phenotypic variation with qERN8-1 contributing the highest percentage in any environment. The total phenotypic variation explained by all identified QTLs ranged from 40.03 to 48.30 %. Five of the 12 QTLs were detected across three environments, and the other seven QTLs were location-specific QTLs. Notably, three of the five QTLs, qERN2-1, qERN8-1 and qERN8-2, were consistently identified in each environment, and the ERN variation explained by them ranged from 24.92 % for 13YN to 37.36 % for 12YN. In addition, the positive alleles of all the 12 QTLs indicated that B73 had a positive effect on increasing the ERN.

Molecular linkage map of RIL and locations of QTL for ERN and TR across multiple environments. The letter ‘C’ means chromosome. The QTLs for each trait in each environment are differentiated by lines with different colors. The traits attached environments represent the QTLs detected in Yunnan in 2012, Yunnan in 2013, Sichuan in 2014, Neimenggu in 2014, respectively. The linkage map was produced by the Mapchart 2.2 software (http://www.wageningenur.nl/en/show/Mapchart.htm). (Color figure online)
Eight putative QTLs for TR were observed in the present study and located on chromosome 1, 2, 3, 4, 6 and 8. The phenotypic variation explained by individual QTL ranged from 2.09 (qTR4-1) to 12.99 % (qTR8-1). Together, those detected QTLs explained 23.57–33.78 % of TR variation across different environments. Among them, five QTLs were repeatedly identified in multiple environments, and the rest QTLs were environment-specific. The negative additive effect of all QTLs indicated that alleles came from SICAU1212 except one location-specific QTL, qTR3-1. Interestingly, two of the five QTLs (qTR8-1 and qTR8-2) basically had stronger effects on TR, regardless of the environment. Remarkably, one QTL, qTR2-1 mapped in the same marker interval chr2-zfl2–chr2-13000, was associated with the TR across all environments explaining 4.27–4.92 % of the phenotypic variation. It was noteworthy that these three QTLs and two location-specific QTLs (qTR1-2 and qTR4-1) were detected in the same map position with QTLs for the ERN. In a word, five common QTLs, located within chr1-90229–chr1-100909, chr2-zfl2–chr2-13000, chr4-203129–chr4-213371, chr8-18426–chr8-19676 and chr8-103366–chr8-111393, may simultaneously control ERN and TR.
The joint QTL and QTL environment interaction
Thirteen and ten significant QTLs were mapped for ERN and TR by joint analysis, respectively (Supplementary Table S2). Ten of the 13 and four of the ten QTLs were consistent with those of ERN and TR through single environment mapping, respectively. Each significant ERN QTL with additive effect explained from 0.01 to 0.27 of phenotypic variance. For TR, the phenotypic variance contributed by individual QTL with additive effect varied between 0.001 and 0.24. The total phenotypic variances of ERN and TR explained by respective QTLs were the same (0.87). Only one QTL, qERN8-1, was involved in significant QTL × environment interaction and represented additive by environment interaction effect in this study. And 0.71 % of the heritability was accounted for by this QTL.
Epistatic interaction among identified QTLs
A total of nine pairs of significant epistatic interactions (two for ERN and seven for TR) were detected with additive by additive interaction among all the four environments (Table 5). These involved 12 QTLs dispersed on chromosomes 1, 2, 3 and 8. Eight of 9 epistatic interactions existed within significant QTLs detected by joint analysis, while the rest occurred between a significant QTL and a non-significant QTL located on chromosome 2. The epistatic interaction between qTR3-1 and qTR-J8-(3) detected in 13YN explained the largest heritability of the additive by additive epistatic interaction and had the decreasing effect on TR. One epistatic interaction between qTR2-1 and qTR8-1 was repeatedly identified in all environments with the exception of 12YN, indicating that this epistasis consistently affected the genetic basis of TR. In addition, two genomic regions simultaneously influencing ERN and TR within chr2-zfl2–chr2-13000 and chr8-16745–chr8-18426, were identified as involved in five and four epistasis, respectively. Although several epistasis were detected for the two traits, their effects were all less lower than those of the corresponding QTLs, indicating that the main effect of significant QTL may have stronger effect on ERN and TR.
Discussion
The domestication process from teosinte to maize may be influenced by three major mutations. The first mutation was that the glumes of teosinte kernels changed from hardened to softening, which could increase the accessibility of kernels for harvest (or liberation); the second was that the pedicellate spikelets did not abort early in development; and the third was that the value of ERN varied from four to more (Iltis 2000). The occurrence of two-ranked specimens supported the above mentioned hypothesis (Benz 2001). After domestication, wild four-rowed maize with two ranks was the extreme case, whereas modern maize forms approximately 8–20 rows with many ranks. A four-rowed maize material P4 derived from an eight-rowed variety was made to analysis the inheritance of the ERN in maize (Daniel 1963). Interestingly, SICAU1212 exhibited two ranks with two rows per rank (Fig. 1), and its progenitor, silunuo, had many primordial traits similar to teosinte (two ranks with one rows per rank) (Zeng et al. 1981). Some studies showed that the alleles that increase the ERN originated equally from both parents (Li et al. 2007, 2011; Upadyayula et al. 2006; Veldboom and Lee 1994), on the contrary, any decreasing effects of QTLs on the ERN were attributed to the parent (SICAU1212) with the lower ERN in this study, which agrees with the findings obtained from maize × teosinte segregating populations (Briggs et al. 2007; Doebley and Stec 1991, 1993) and those from a part of maize × maize segregations (Cai et al. 2014; Tian et al. 2014; Yu et al. 2014). One possible reason elucidating the above phenomenon is that there is not more variation on ERN within modern maize germplasms, namely due to limited variation in the ERN. The missing genetic variation from 4 to more rows could not be determined, although MT-6 with an ERN of six was crossed with B73 to produce different populations (Cai et al. 2014). Thus, SICAU1212 × B73 population made to detect the QTL for ERN and TR could increase the phenotypic difference between the parents, further gain some genetic basis underlying ERN during maize subsequent improvement and provide some effective information about the TR inheritance in maize.
Genetic architectures of ERN and TR
Segregations between maize and teosinte were made to identify ERN QTL associated with domestication, while diverse maize populations were suitable for detection of QTL affecting diversification. The first consistent QTL in this study, qERN2-1, was also located on bin 2.02. Multiple studies have shown that this region on the short arm of chromosome 2 had a large effect on ERN (Cai et al. 2014; Doebley and Stec 1991, 1993; Karen Sabadin et al. 2008), indicating a potential QTL region near qERN2-1 may play a novel role in both maize domestication and later improvement processes. Both the second and third consistent QTLs (qERN8-1 at bin 8.02 and qERN8-2 at bin 8.04) were located on chromosome 8. The former QTL was differentiated from the latter one by having larger effect on ERN. Previous reports of QTL for ERN identified in the two genomic regions in other maize populations were rather sparse. Briggs et al. (2007) reported that a chromosomal region near qERN8-1 influenced ERN in a large BC1 population derived from maize and teosinte. One minor QTL near qERN8-2 explaining 6 % of ERN variation has been identified (Doebley 1992; Doebley et al. 1990). These implied that qERN8-1 and qERN8-2 may be domestication-related QTLs those control the initial switch from four to more rows of kernel.
In addition to the three consistent QTLs, other two QTLs (qERN4-3 at bin 4.08 and qERN5-1 at bin 5.04) identified in only three environments also influenced the genetic architecture of ERN. An interesting case is the QTL detected on bin 4.08, which has been repeatedly reported to significantly affect ERN (Austin and Lee 1996; Li et al. 2014a; Veldboom and Lee 1994, 1996; Yan et al. 2006) and also contained QTLs for other agronomic traits, such as grain yield (Lu et al. 2006), tassel branch number (Chuck et al. 2014), ear diameter (Li et al. 2011), kernel number per row (Lu et al. 2011), kernel weight, width and thickness (Liu et al. 2014). Therefore, bin 4.08 is noteworthy for genetic improvement of morphological traits in maize. Similarly, region near qERN5-1 was also frequently reported to be involved in the QTL for ERN in diverse populations (Li et al. 2007, 2009; Szabó and Burr 1996; Tian et al. 2014). By 259 highly homozygous NIRILs, one QTL accounting for 53.4 % of ERN variation was mapped to approximately 168 Mb (physical position) on chromosome 5 (Lemmon and Doebley 2014). Collectively, there are high probabilities that genes underlying ERN variation change exist in the five regions. High-density SNP arrays and DNA re-sequencing have supplied more comprehensive information of the genotypes of plants and animals. GWAS aims to detect variants at candidate loci or genes for traits. Notably, eight of the 12 QTLs for ERN detected in single environment in our study had the coincident marker intervals with those of GWAS (physical distance of two <10 Mb) (Brown et al. 2011). It seems that the location of QTL for ERN in this study had a high reliability.
Recently, K10HEX206 with four rows also derived from silunuo has been employed to dissect the genetic improvement of ERN (Jiao et al. 2014). Of the 12 QTLs identified by them in 210 F2:3 families genotyped by 173 molecular markers, only six QTLs exhibited coincidence with those of the present study, possibly due to the different genetic background, population size and marker densities. On the one hand, RIL raised the recombinant frequency between linked loci and presented the stronger power for the identification of QTL compared with the F2 population (Cowen 1988). On the other hand, the lack of a moderate segregation population size affected the consistency of QTL detection (Melchinger et al. 1998) and estimates of the allelic richness similarly (Kalinowski 2004, 2005).
As noted earlier, TR was considered to be one key trait distinguishing maize and teosinte (Doebley 2004). In an overview of QTL distribution in the previous reports (Doebley and Stec 1991; Doebley et al. 1990; Langham 1940; Mangelsdorf 1947; Rogers 1950; Szabó and Burr 1996), QTL for TR were located on all chromosomes with the exception of chromosome 7. In this study, QTLs involved in inheritance of TR were identified on all chromosomes except for chromosomes 7, 9 and 10 based on the results of single environment mapping and joint analysis. A QTL region (or tr1 gene) with the strongest on the segment of 2S has been detected for this trait (Doebley and Stec 1991), which was consistent with our finding that a region near marker chr2-zfl2 showed stable association with TR. However, our results revealed that the QTL with strongest effect on TR was located on chromosome 8 not 2, which also disagreed with those of previous studies (Doebley 1992; Doebley and Stec 1993).
Several explanations may be advanced to account for the discrepancies between the above researches. The first concerned the different measurement of TR prior to the analysis of its inheritance. Some researchers performed similar Mendelian analyses, with exploring its inheritance by fitting quantitative variants into discrete categories and omitting the mixed ranks (two-ranked on the top whereas many-ranked on the basal). However, mixed phenotype occurred not only in natural maize-teosinte hybrids but also in ears of a high-altitude Toluca teosinte (Orr et al. 2002). We also have attempted to make two- (the TR value ≥ 25 %) versus many-ranked (the TR value < 25 %) ears approach an approximately 1:3 ratio. The result showed that four QTLs (qTR1-1, qTR2-1, qTR8-1 and qTR8-2) had significant associations with the inheritance of TR during four environments (Supplementary table S3), indicating that the inheritance of TR was always controlled by polygenic model, whichever phenotypic measurement was performed, which agreed with Iltis’ views (1983), namely the inheritance of the morphological differences between maize and teosinte was polygenic. Additionally, the lack of the limited number of linkage markers influenced the power of QTL identification. Most investigators used few morphological markers per chromosome, resulting in a part of genomic region may not be adequately covered. The probability of getting false positive would also rise while a relatively larger number of markers genotyped a small number of individuals (Szabó and Burr 1996). These possibilities were minimized as the interval between any two markers in one chromosome was approximately 10 Mb in the present report. Thirdly, genetic background (SICAU1212) used to study the inheritance of TR in this study was quite different from those of the previous researches (teosinte used). As mentioned earlier, the progenitor of SICAU1212, silunuo, exhibited many primordial traits similar to teosinte (Zeng et al. 1981), and it might be derived from normal maize under special ecological environments (Tian et al. 2009), but two key issues are (1) SICAU1212 is not really a teosinte; (2) whether the molecular mechanism underlying the TR was the same under different genetic backgrounds? Thus, we used SICAU1212 as a parent so that we could detect the TR loci or genes involved in maize improvement rather than those associated with maize domestication. In conclusion, QTL region on chromosome 2 seems to mainly contribute to TR during domestication, whereas QTL region on chromosome 8 seems to be mainly responsible for lateral improvement steps.
Epistasis between QTL
Epistasis, or interactions between genes may play important roles in understanding the genetic basis of complex quantitative traits (Phillips 2008). In this study, the total phenotypic variation explained by all significant QTLs was always less than the broad-sense heritability, regardless of the trait and environment, indicating that the missing proportion of phenotypic variation may be partly explained by epistasis (Carlborg and Haley 2004). For ERN, two pairs of significant epistatic interactions have been detected in our report, mostly among significant QTLs, indicating that the inheritance of ERN was controlled by few major genes plus several minor genes and epistatic effects. Similar results were previously observed (Li et al. 2009; Yan et al. 2006). Notably, one stable epistasis involved in region near qERN2-1 has been found in different environments (Cai et al. 2014). More recently, Sa et al. (2015) showed that genetic interactions involved in regions near qERN8-1 or qERN8-2 were associated with ear height, stem thickness or grain yield. Additionally, one pair of epistatic interaction involved in region near qERN8-2 controlling kernel length was reported by Zhang et al. (2014). Therefore, the three QTL regions may be considered as epistatic regulators, which act on the development of maize.
Although dozens of QTLs controlling TR have been detected in diverse maize-teosinte hybrids, no epistasis for this trait has been reported. One stable epistatic interaction (qTR2-1/qTR8-1) was identified in our study. It was noteworthy that qTR2-1 has tr1 gene as flanking marker. Similar to a strong epistatic effect between teosinte branched1 (tb1) and one QTL on chromosome arm 3L occurred, other epistasis for other key traits controlling the evolution of maize could exist. These epistatic interactions may indirectly affect the developmental processes (Doebley 2004). Thus, the cryptic region near qTR8-1 or qERN8-1 may arouse more experts’ interest.
Mutant genes and the formation of the ERN and TR
The ERN is considered one of the key traits selected by humans during maize domestication and an obviously unique characteristic of maize ear development. If one QTL region related with certain trait contained or was adjacent to a known mutant gene, which was the only candidate gene affecting the development of this trait, the mutant gene may be regarded as a logical candidate gene (Robertson 1985; Upadyayula et al. 2006).
In the present study, one stable QTL (qERN8-1) near 18 Mb (based on the AGI’s B73 RefGen_v2 sequence) simultaneously influenced the ERN and TR. Two mutant genes (vt2 and bif1) mapped to approximately 17 and 22 Mb on chromosome 8, respectively, are possible candidate genes in this region. The vt2 mutant ear typically had barren patches on one or both sides of the ear (Phillips et al. 2011) and this phenotype was similar to that of SICAU1212 (Fig. 1). The mutant gene bif1 played a role in auxin transport and may make ear exhibit an intermediate (two/more-ranked) ear (phyllotaxy), similar to the characteristic of TR (Barazesh and McSteen 2008). Another QTL (qERN2-1) on chromosome 2S responsible for both ERN and TR has zfl2 gene as flanking marker. The zfl2 gene was not only contributed to changes in ERN during maize domestication but also associated with further improvement of ERN during the diversification stage (Bomblies and Doebley 2006; Bomblies et al. 2003; Doebley 2004). Maize SBP-box transcription factor gene, ub3, which affected the rate of cell differentiation to the lateral domains of meristems and directly influenced yield (Chuck et al. 2014), was located approximately 199 Mb on chromosome 4 near the QTL qERN4-3. One environment-specific QTL for ERN, qERN4-1, located approximately 4 Mb on chromosome 4 contain sos1 gene. Sos1 caused a reduction in the number of tassel branch and ERN (Doebley et al. 1995). Ramosa1 enhancer locus2 (rel2) was co-localized with another location-specific QTL (qERN10-1). Rel2 physically interacted with ramosa1, resulting in the determinacy of spikelet-pair meristems (Gallavotti et al. 2010). Many of the mutant genes affecting ear development have not yet been mapped across B73 × SICAU1212 segregation. One reason for this lack may be that these genes do not control the ERN diversity between B73 and SICAU1212. Further studies are needed to determine whether these candidate genes are the causal genetic variants for these QTLs.
Common QTL
The ultimate objective of mapping the QTL for certain trait is to dissect the molecular and genetic basis of its variation. Meanwhile, the identification and confirmation of QTL that simultaneously influences several agronomic traits (common QTL or QTL with pleiotropic effect) could offer greater opportunities for plant breeding. For example, gene controlling grain chalkiness in rice, Ckalk5 (Li et al. 2014b), was located within the region simultaneously associated with grain width and length–width ratio in an earlier report (Tan et al. 2000); GS3 in rice, which was regarded as a major QTL for grain length, weight, width and thickness (Fan et al. 2006), could vary grain size by modifying four putative domains (Mao et al. 2010); in maize, one QTL on chromosome 10 that had pleiotropic effect on kernel number per row (KN), ERN and ear diameter in high and low phosphorus treatments has been fine mapping for KN (Li et al. 2010; Zhang et al. 2013).
Three common QTLs detected in this report simultaneously controlled ERN and TR due to the utilization of SICAU1212 with four rows. Although SICAU1212 may not be an appropriate breeding base, this research could well aid in understanding the genetic basis of these traits and provide a foundation for further fine-mapping of these common QTLs.
References
Austin DF, Lee M (1996) Comparative mapping in F2:3 and F6:7 generations of quantitative trait loci for grain yield and yield components in maize. Theor Appl Genet 92:817–826
Barazesh S, McSteen P (2008) Barren inflorescence1 functions in organogenesis during vegetative and inflorescence development in maize. Genetics 179:389–401
Beavis WD, Smith OS, Grant D, Fincher R (1994) Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci 34:882–896
Benz BF (2001) Archaeological evidence of teosinte domestication from Guilá Naquitz, Oaxaca. Proc Natl Acad Sci 98:2104–2106
Bomblies K, Doebley JF (2006) Pleiotropic effects of the duplicate maize FLORICAULA/LEAFY genes zfl1 and zfl2 on traits under selection during maize domestication. Genetics 172:519–531
Bomblies K, Wang R-L, Ambrose BA, Schmidt RJ, Meeley RB, Doebley J (2003) Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 130:2385–2395
Bommert P, Nagasawa NS, Jackson D (2013) Quantitative variation in maize kernel row number is controlled by the FASCIATED EAR2 locus. Nat Genet 45:334–337
Briggs WH, McMullen MD, Gaut BS, Doebley J (2007) Linkage mapping of domestication loci in a large maize–teosinte backcross resource. Genetics 177:1915–1928
Brown PJ, Upadyayula N, Mahone GS, Tian F, Bradbury PJ, Myles S, Holland JB, Flint-Garcia S, McMullen MD, Buckler ES (2011) Distinct genetic architectures for male and female inflorescence traits of maize. PLoS Genet 7:e1002383
Cai L, Li K, Yang X, Li J (2014) Identification of large-effect QTL for kernel row number has potential for maize yield improvement. Mol Breed 34:1087–1096
Carlborg Ö, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
Choe E, Rocheford TR (2012) Genetic and QTL analysis of pericarp thickness and ear architecture traits of Korean waxy corn germplasm. Euphytica 183:243–260
Chuck GS, Brown PJ, Meeley R, Hake S (2014) Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc Natl Acad Sci 111:18775–18780
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Collard BC, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572
Cowen N (1988) The use of replicated progenies in marker-based mapping of QTL’s. Theor Appl Genet 75:857–862
Daniel L (1963) Analysis of inheritance of the number of kernel rows in maize. Theor Appl Genet 33:290–301
Darvasi A, Soller M (1997) A simple method to calculate resolving power and confidence interval of QTL map location. Behav Genet 27:125–132
Doebley J (1992) Mapping the genes that made maize. Trends Genet 8:302–307
Doebley J (2004) The genetics of maize evolution. Annu Rev Genet 38:37–59
Doebley J, Stec A (1991) Genetic analysis of the morphological differences between maize and teosinte. Genetics 129:285–295
Doebley J, Stec A (1993) Inheritance of the morphological differences between maize and teosinte: comparison of results for two F2 populations. Genetics 134:559–570
Doebley J, Stec A, Wendel J, Edwards M (1990) Genetic and morphological analysis of a maize-teosinte F2 population: implications for the origin of maize. Proc Natl Acad Sci 87:9888–9892
Doebley J, Stec A, Kent B (1995) Suppressor of sessile spikelets 1 (Sos1): a dominant mutant affecting inflorescence development in maize. Am J Bot 82:571–577
Dofing S, ĎCroz-Mason N, Thomas-Compton M (1991) Inheritance of expansion volume and yield in two popcorn × dent corn crosses. Crop Sci 31:715–718
Edwards M, Stuber C, Wendel J (1987) Molecular-marker-facilitated investigations of quantitative-trait loci in maize. I. Numbers, genomic distribution and types of gene action. Genetics 116:113–125
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Gallavotti A, Long JA, Stanfield S, Yang X, Jackson D, Vollbrecht E, Schmidt RJ (2010) The control of axillary meristem fate in the maize ramosa pathway. Development 137:2849–2856
Guo J, Su G, Zhang J, Wang G (2008) Genetic analysis and QTL mapping of maize yield and associate agronomic traits under semi-arid land condition. Afr J Biotechnol 7:1829–1838
Hallauer AR, Miranda J (1988) Quantitative genetics in maize breeding, 2nd edn. Iowa State Univ Press, Ames
Iltis HH (1983) From teosinte to maize: the catastrophic sexual transmutation. Science 222:886–894
Iltis HH (2000) Homeotic sexual translocations and the origin of maize (Zea mays, Poaceae): a new look at an old problem. Econ Bot 54:7–42
Jiao F, Li Y, Chen L, Liu Z, Shi Y, Song Y, Zhang D, Li Y, Wang T (2014) Genetics dissection for kernel row number in the specific maize germplasm four-rowed waxy corn. Sci Agric Sin 47:1256–1264
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543
Kalinowski ST (2005) hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Karen Sabadin P, de Souza Lopes, Júnior C, Pereira de Souza A, Augusto Franco Garcia A (2008) QTL mapping for yield components in a tropical maize population using microsatellite markers. Hereditas 145:194–203
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Langham DG (1940) The inheritance of intergeneric differences in Zea-Euchlaena hybrids. Genetics 25:88–107
Lauter N, Doebley J (2002) Genetic variation for phenotypically invariant traits detected in teosinte: implications for the evolution of novel forms. Genetics 160:333–342
Lemmon ZH, Doebley JF (2014) Genetic dissection of a genomic region with pleiotropic effects on domestication traits in maize reveals multiple linked QTL. Genetics 198:345–353
Leng ER (1963) Component analysis in inheritance studies of grain yield in maize. Crop Sci 3:187–190
Li Y, Niu S, Dong Y, Cui D, Wang Y, Liu Y, Wei M (2007) Identification of trait-improving quantitative trait loci for grain yield components from a dent corn inbred line in an advanced backcross BC2F2 population and comparison with its F2:3 population in popcorn. Theor Appl Genet 115:129–140
Li H, Ribaut J-M, Li Z, Wang J (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116:243–260
Li Y, Li X, Li J, Fu J, Wang Y, Wei M (2009) Dent corn genetic background influences QTL detection for grain yield and yield components in high-oil maize. Euphytica 169:273–284
Li M, Guo X, Zhang M, Wang X, Zhang G, Tian Y, Wang Z (2010) Mapping QTLs for grain yield and yield components under high and low phosphorus treatments in maize (Zea mays L.). Plant Sci 178:454–462
Li J, Zhang Z, Li Y, Wang Q, Zhou Y (2011) QTL consistency and meta-analysis for grain yield components in three generations in maize. Theor Appl Genet 122:771–782
Li F, Jia H, Liu L, Zhang C, Liu Z, Zhang Z (2014a) Quantitative trait loci mapping for kernel row number using chromosome segment substitution lines in maize. Genet Mol Res 13:1707–1716
Li Y, Fan C, Xing Y, Yun P, Luo L, Yan B, Peng B, Xie W, Wang G, Li X (2014b) Chalk5 encodes a vacuolar H+-translocating pyrophosphatase influencing grain chalkiness in rice. Nat Genet 46:398–404
Lincoln SE (1992) Mapping genes controlling quantitative traits with MAPMAKER/QTL 1.1. Whitehead Institute Technical Report
Liu X, He S, Zheng Z, Huang Y, Tan Z, Wu X (2010) QTL identification for row number per ear and grain number per row in maize. Maydica 55:127–133
Liu Y, Wang L, Sun C, Zhang Z, Zheng Y, Qiu F (2014) Genetic analysis and major QTL detection for maize kernel size and weight in multi-environments. Theor Appl Genet 127:1019–1037
Lu GH, Tang JH, Yan JB, Ma XQ, Li JS, Chen SJ, Ma JC, Liu ZX, Zhang YR, Dai JR (2006) Quantitative trait loci mapping of maize yield and its components under different water treatments at flowering time. J Integr Plant Biol 48:1233–1243
Lu M, Xie C-X, Li X-H, Hao Z-F, Li M-S, Weng J-F, Zhang D-G, Bai L, Zhang S-H (2011) Mapping of quantitative trait loci for kernel row number in maize across seven environments. Mol Breed 28:143–152
Mangelsdorf PC (1947) The origin and evolution of maize. Adv Genet 1:161–207
Mao H, Sun S, Yao J, Wang C, Yu S, Xu C, Li X, Zhang Q (2010) Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci 107:19579–19584
Melchinger AE, Utz HF, Schön CC (1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149:383–403
Orr AR, Mullen K, Klaahsen D, Sundberg MD (2002) Inflorescence development in a high-altitude annual Mexican teosinte (Poaceae). Am J Bot 89:1730–1740
Petrovic Z (1998) Combining abilities and mode of inheritance of yield and yield components in maize (Zea mays L.). NoviSad 8:81–85
Phillips PC (2008) Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9:855–867
Phillips KA, Skirpan AL, Liu X, Christensen A, Slewinski TL, Hudson C, Barazesh S, Cohen JD, Malcomber S, McSteen P (2011) vanishing tassel2 encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize. Plant Cell 23:550–566
Qi Z, Wu Q, Han X, Sun Y, Du X, Liu C, Jiang H, Hu G, Chen Q (2011) Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 179:499–514
Robertson DS (1985) A possible technique for isolating genic DNA for quantitative traits in plants. J Theor Biol 117:1–10
Rogers JS (1950) The inheritance of inflorescence characters in maize-teosinte hybrids. Genetics 35:541–558
Sa KJ, Park JY, Woo SY, Ramekar RV, Jang C-S, Lee JK (2015) Mapping of QTL traits in corn using a RIL population derived from a cross of dent corn × waxy corn. Genes Genom 37:1–14
Saghai-Maroof M, Soliman K, Jorgensen RA, Allard R (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci 81:8014–8018
Santos FR, Pena SD, Epplen JT (1993) Genetic and population study of a Y-linked tetranucleotide repeat DNA polymorphism with a simple non-isotopic technique. Hum Genet 90:655–656
Sreckov Z, Nastasic A, Bocanski J, Djalovic I, Vukosavljev M, Jockovic B (2011) Correlation and path analysis of grain yield and morphological traits in test–cross populations of maize. Pak J Bot 43:1729–1731
Stuber CW, Edwards MD, Wendel JF (1987) Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Sci 27:639–648
Szabó VM, Burr B (1996) Simple inheritance of key traits distinguishing maize and teosinte. Mol Gen Genet 252:33–41
Taguchi-Shiobara F, Yuan Z, Hake S, Jackson D (2001) The fasciated ear2 gene encodes a leucine-rich repeat receptor-like protein that regulates shoot meristem proliferation in maize. Genes Dev 15:2755–2766
Tan Y, Xing Y, Li J, Yu S, Xu C, Zhang Q (2000) Genetic bases of appearance quality of rice grains in Shanyou 63, an elite rice hybrid. Theor Appl Genet 101:823–829
Tang J, Yan J, Ma X, Teng W, Wu W, Dai J, Dhillon BS, Melchinger AE, Li J (2010) Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL mapping in an immortalized F2 population. Theor Appl Genet 120:333–340
Tian M, Tan G, Liu Y, Rong T, Huang Y (2009) Origin and evolution of Chinese waxy maize: evidence from the Globulin-1 gene. Genet Resour Crop Evol 56:247–255
Tian B, Wang J, Wang G (2014) Confirmation of a major QTL on chromosome 10 for maize kernel row number in different environments. Plant Breed 133:184–188
Upadyayula N, Da Silva H, Bohn M, Rocheford T (2006) Genetic and QTL analysis of maize tassel and ear inflorescence architecture. Theor Appl Genet 112:592–606
Veldboom LR, Lee M (1994) Molecular-marker-facilitated studies of morphological traits in maize. II: determination of QTLs for grain yield and yield components. Theor Appl Genet 89:451–458
Veldboom LR, Lee M (1996) Genetic mapping of quantitative trait loci in maize in stress and nonstress environments: I. Grain yield and yield components. Crop Sci 36:1310–1319
Yan J, Tang H, Huang Y, Zheng Y, Li J (2006) Quantitative trait loci mapping and epistatic analysis for grain yield and yield components using molecular markers with an elite maize hybrid. Euphytica 149:121–131
Yang J, Zhu J, Williams RW (2007) Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics 23:1527–1536
Yu Y, Li G, Yang Z, Hu J, Zheng J, Qi X (2014) Identification of a major quantitative trait locus for ear size induced by space flight in sweet corn. Genet Mol Res 13:3069–3078
Zeng M, Yang T, Wang P (1981) The relative analyses of maize cultivar menghai four-row wax. Acta Genet Sin 8:91–96
Zhang G, Wang X, Wang B, Tian Y, Li M, Nie Y, Peng Q, Wang Z (2013) Fine mapping a major QTL for kernel number per row under different phosphorus regimes in maize (Zea mays L.). Theor Appl Genet 126:1545–1553
Zhang Z, Liu Z, Hu Y, Li W, Fu Z, Ding D, Li H, Qiao M, Tang J (2014) QTL analysis of kernel-related traits in maize using an immortalized F2 population. PLoS ONE 9:e89645
Zhou J, Fang H, Shan J, Gao X, Chen L, Xie C, Xie T, Liu J (2014) A major QTL located on chromosome V associates with in vitro tuberization in a tetraploid potato population. Mol Genet Genomics 289:575–587
Acknowledgments
We greatly appreciate Dr. Wenming Wang, Zhiming Zhang, Jihua Tang, Yaou Shen, Yanli Lu and Yuanqi Wu for suggestions and revisions to this manuscript. This research was supported by the National Basic Research Program of China (the “973” project, 2014CB138203) and National Natural Science Foundation of China (31101161).
Conflict of interest
All of the authors declare that they have no conflict of interest.
Ethical standards
The experiments comply with the current laws of the country in which they were performed.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, C., Tang, D., Zhang, L. et al. Identification of QTL for ear row number and two-ranked versus many-ranked ear in maize across four environments. Euphytica 206, 33–47 (2015). https://doi.org/10.1007/s10681-015-1466-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-015-1466-4