
Abstract
Ear traits play a vital role in maize (Zea mays) yield. The ear angle (EA) has an obvious impact on corn yield and planting density as well as other maize ear traits. However, the genetic control of EA is still unclear. In this study, we identified quantitative trait loci (QTLs) for EA from four recombinant inbred line populations, BY815/DE3, BY815/K22, CI7/K22 and Mo17/X26-4, which were grown in three environments, and the genetic architecture of EA in maize was subsequently dissected. The results indicated that maize EA was highly heritable and was affected by both genotype and environment. Based on the genetic linkage map constructed using 56,110 bins as markers, nine effective QTLs for maize EA were detected locating on chromosomes 2, 3, 4, 6 and 7. These QTLs accounted for different EA variations ranging from 5.5% (qCIKEA6) to 7.6% (qBYKEA3). Moreover, 14 candidate genes were identified from the reduced QTLs using a bin-map method, which mainly encoded enzymes in signal transduction, transcriptional regulation and metabolism. Conclusively, our results can benefit further study of the genetic basis of EA and improve the maize EA quality through molecular breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nowadays, maize (Zea mays L.) is a staple food crop worldwide as a starch source as well as a raw material in the feed and energy industries (Wang et al. 2015). With a growing world population and demand, increasing maize yield is an effective method to meet the requirement (James and Myers 2009). Dehydration rate of maize grains affects mechanical harvest and storage, while maize ear angle (EA) has a direct and substantial influence on dehydration and maize yield (Zhang et al. 2012). In addition, Tang and Ding (2008) found that smaller maize EAs affected pollination, which resulted in a lower seed-setting percentage and finally decreased the maize yield. Sun (2019) reported that maize EAs get smaller with increased planting density. Hence, to illustrate the genetic basis for maize EA and to improve the EA trait will definitely benefit maize breeding.
Maize ears are the main harvested organs and ear traits directly impact the yield. Robinson et al. (1951) proved that it is more efficient to breed high-quality maize by selecting ear traits as yield traits. A variety of maize ear traits, including ear length, ear weight and ear line number, have been noted to possess significant correlations with maize yield (Li et al. 2016a, b; Tong 2015; Li et al. 2010; Song et al. 2006). Recent studies showed that ear tip-barrenness and ear rot resistance could also influence maize yield, and their genetic architectures were investigated (Li et al. 2020; Guo et al. 2020). Among all ear traits, little is known about maize EA. However, previous studies have revealed that rice panicle angle (PA), which is identical to maize EA, was obviously related to rice yield. Liu and Hong (2005) found that PA affected not only rice yield but also rice quality. Jiang et al. (2007) stated that rice PA shared positive correlations with grain length, the grain length/width ratio, and the milled rice rate. Moreover, the genetic basis for rice PA and potential candidate genes have also been investigated (Chen et al. 2006; Niu et al. 2013). In addition, a latest report showed that the panicle angle of Tef [Eragrostis tef (Zucc.) Trotter] contributed to the likelihood of lodging tolerance (Blösch et al. 2020). All those imply that the maize EA is an important ear trait which might have a potential influence on other maize ear traits and maize yield.
As a classic method, quantitative trait loci (QTL) mapping is widely applied to identify loci contributing to specific quantitative traits without a prior genetic background (Goldman et al. 1993). Plenty of QTLs for maize ear traits have been identified to date (Veldboom and Lee 1994; Guo et al. 2008; Tang et al. 2010; Wang et al. 2016; Huang et al. 2016). For example, Correlations and heritability for several maize ear fasciation-related traits and yield were determined by Mendes-Moreira et al. (2015) using a segregating population. In addition, QTLs associated with maize leaf angle have been extensively reported (Ding et al. 2015; Li et al. 2015; Zhang et al. 2014; Ku et al. 2010). Ten QTLs related to maize leaf angle were detected from the B73/Mo17 RIL population in two locations, among which one large effect QTL accounted for 27.7 variation (Mickelson et al. 2002). Furthermore, Niu et al. (2013) determined eight QTLs for PA in a 254 rice recombinant inbred line using SSR markers. Similar to QTLs for maize leaf angle and rice panicle angle, punctual maize EA QTLs need to be identified for further investigation. In addition, with the development of genomics and genotyping technologies, high-density single nucleotide polymorphism (SNP) markers have been utilized to increase marker density for their low-cost, time-saving and high-throughput properties (Van et al. 2015). Using saturated SNP markers and combining co-segregating markers into one bin and separating adjacent bins on the basis of a single recombinant event, we can successfully examine most recombination events and determine narrower QTL intervals (Zou et al. 2012; Orawan et al. 2017; Li et al. 2016a, b; Song et al. 2016).
The objectives of our study were (1) to illustrate the genetic basis of maize EA by refining the position of identified QTLs, (2) to screen for the candidate genes responsible for maize EA, and (3) to dissect the genetic architecture of maize EA. Our expectation is that the results obtained from this study will be helpful in improving maize yield and quality.
Materials and methods
Maize populations
Four maize RIL populations were kindly supplied by Professor Yang (National Maize Improvement Centre of China, China Agricultural University, Beijing). The BYD (F6), BYK (F6), CIK (F7) and MX (BC2F5) populations were derived from the cross between the inbred lines BY815/DE3, BY815/K22, CI7/K22 and MO17/X26-4, respectively. BY815 is an inbred line developed from a Beijing high-oil population, and K22 is a Chinese elite inbred line derived from a cross between the two Chinese inbred lines, LK11 and Ye478 (Yang et al. 2010). CI7 is a high-carotenoid and late maturing line introduced from America. DE3 was derived from a Germplasm Enhancement of Maize breeding cross (Xiao et al. 2016). The BC2F5 population was derived from a cross between Mo17 and a wild teosinte X26-4 (Z. mays ssp. Mexicana, 566686) (Pan et al. 2016).
Growth and phenotyping
Four maize RIL populations, including 207 BYD lines, 207 BYK lines, 196 CIK lines and 191 MX lines, along with their respective parents, were grown at three locations in China, Beijing (40°08′N, 116°10′E), Neimeng (40°31′N, 107°05′E) and Liaoning (41°82′N, 123°56′E) in 2015. All the lines were planted in a single-row blot which was conducted with one replication under a completely randomized block design. More than five plants in each row were self-pollinated in order to obtain homozygous offspring. On the 15th day after pollen emission, the EAs were measured using an independent R&D angle detector which is a smart steel parallelogram with a degree display (Fig. 1). When measuring EAs, we parallel the two arms opposite to the display to the inner side of the maize ear and the maize stem, respectively, and read the angle degree on the display, as the opposite angles in a pair are equal in a parallelogram. Each angle degree was recorded to the decimal point. The data of four maize ear traits, including ear length (EL), ear row number (ERN), ear weight (EW) and cob weight (CW), were kindly shared from a previous study (Xiao et al. 2016). Pearson correlation coefficients were calculated for comparison of maize EA with these ear traits according to the method from Tai et al. (2016).

An independent R&D EA detector used in this study to measure maize EAs
Statistical analysis
All statistics were analyzed and performed via R software 3.1.1 (www.R-project.org). A mixed linear model was fitted to obtain the best linear unbiased prediction (BLUP) for maize EA. The model for BLUP was: yi = µ + fi + ei + εi, where yi is the individual phenotypic value, μ is the grand mean for all environments, fi is the genetic effect, ei is the effect of different environments, and εi is the random error. The grand mean was considered as a fixed effect, and genotype and environment were fitted as random effects. An ANOVA of all maize EAs in different RIL populations was performed using the aov function in R software to estimate different variance components, including VG (genetic variance), VE (environmental variance), VP (total phenotypic variance), VA (additive genetic variance) and VNA (non-additive genetic variance), where VP is the sum of VG and VE, and VG is the sum of VA and VNA. Then, those variance components were employed to calculate the broad-sense heritability as: H2 = VG/VP (Knapp et al. 1985), whereas the narrow-sense heritability was estimated as: h2 = VA/VP, according to the classic method from Falconer (Falconer and Mackay 1996).
Genotyping and the construction of bin and genetic-linkage maps
All 801 lines from four maize RIL populations, together with their respective parents, were genotyped with the Illumina MaizeSNP50 BeadChip (Illumina, San Diego, CA, USA), which contains a total of 56,110 SNPs (Ganal et al. 2011). SNP genotyping was carried out on the Illumina Infinium SNP genotyping platform at the Cornell University Life Sciences Core Laboratories Centre or at DuPont Pioneer (Wilmington, DE, USA). pLink software (Li et al. 2012) was utilized to estimate the missing rate, minor-allele frequency and heterozygosity for each SNP, and the missing rate and heterozygosity for each line.
After quality control, 56,110 SNPs that were polymorphic between each separate parental lines were used to construct the genetic linkage map using an economic go-wrong method with integrated Carthagene software (de Givry et al. 2005) in a Linux system with in-house Perl scripts (www.maizego.org/Resources.html). Co-segregating markers were completely assigned to a chromosomal bin and each bin was considered as one marker (Pan et al. 2016). Details of the bins used to construct genetic linkage maps and recombination dynamics in those RILs and their parent strains have been published previously (Xiao et al. 2016).
QTL mapping
QTL mapping of maize EA was undertaken using composite-interval mapping implemented in Windows QTL Cartographer 2.5 (Basten et al. 2005). The whole genome was scanned with a marker interval of 0.5 cM and a window size of 10 cM. Model 6 of the Zmapqtl module was applied to detect QTLs and to evaluate their effects. A forward–backward stepwise regression with five controlling markers was used to control the background from flanking markers. The threshold LOD values to declare the putative QTLs were estimated via permutation tests with more than 1000 replicates at a significance level of P < 0.05 (Churchill and Doerge 1994). The confidence interval of QTL positions was determined with the 1.5-LOD support interval method (Lander and Botstein 1989). To further detect the interactions between significant QTLs and their total phenotypic variations, multiple interval mapping (MIM) in Windows QTL Cartographer 2.5 was performed using Bayesian Information Criteria (Kao et al. 1999).
Annotation of candidate genes
Based on the available information in the Gramene BioMart database (ensembl.gramene.ort/biomart), genes within the target QTL intervals were extracted. The physical locations of these genes were determined according to the maize genomic sequence v.2. By performing a BLASTP search through the NCBI website (blast.ncbi.nlm.nih.gov/Blast.cgi), the corresponding genes were annotated, and the candidate genes were assigned into various biological processes after gene ontology term analysis.
Results
Phenotypic variation and heritability in maize EA and its correlations with other ear traits
The Best Linear Unbiased Prediction (BLUP) value of EA revealed that the means of the BYD, BYK and MX RIL populations were close to their mid-parent values, whereas the mean EA value of the CIK RIL population was a little higher than the mid-parent value (Table 1). The ANOVA results indicated that there were highly significant effects on the EA trait due to genotypes and environments (Table 1). The EA trait followed normal distributions, as shown in Fig. 2. Moderately positive EA correlations were observed among the three locations in all the four RIL populations, ranging from 0.21 to 0.59 (P < 0.01) (Fig. 2). In addition, the broad-sense heritability estimates showed moderate heritability (H2) for EA in the four RIL populations ranging from 0.48 to 0.63, while the narrow-sense heritability estimates (h2) ranged from 0.32 to 0.50. These indicated the genetic determination of maize EA phenotypic variations, and their suitability for subsequent QTL mapping (Table 1). In addition, correlation analysis showed that maize EA was positively correlated with ERN but negatively correlated with EL, indicating that maize EA was indeed related to some ear traits. However, no significant correlation was observed between maize EA and EW or CW (Fig. 3).

Frequency distributions and correlations of maize EA across different environments in four maize RIL populations. A, B, C and D indicate the BYD, BYK, CIK and MX maize RIL populations, respectively. Bar charts show the phenotypic distribution of maize EA, the values are Pearson correlation coefficients and the corresponding plots are scatter plots of maize EA. BJ, LN and NM represent the environments in Beijing, Liaoning, and Neimeng, respectively. **P ≤ 0.01

Correlation coefficients of maize EA with other maize ear traits based on BLUP value in BYD, BYK and CIK populations. EA ear angle, EL ear length, ERN ear row number, EW ear weight, CW cob weight, EA ear angle. *Significant at P ≤ 0.05
Identification of QTLs for EA
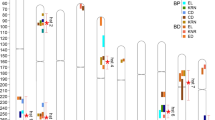
QTL mapping was performed by the composite-interval mapping method (He et al. 2015) to identify QTLs for EA, and phenotypic BLUP values across different environments were adopted to minimize the environmental variation effect. Totally, 56 QTLs associated with maize EA were detected in the four RIL populations, including 12 QTLs from BYD, 13 QTLs form BYK, 16 QTLs from CIK and 15 QTLs from MX, among which 9 QTLs were selected from the BLUP values at empirical threshold LOD values more than 3.0 (Table 2; Fig. 4). These QTLs had an average length of 32.6 Mb (8.4 cM), with a range of 0.6–124.3 Mb (2.3–19.4 cM) which located in nine genomic regions on chromosomes 2, 3, 4, 6 and 7, respectively (Table 2; Fig. 5). In addition, a pair of partial overlapping QTLs, qCIKEA3-1 and qCIKEA3-2 were observed, both of which came from the CIK RIL population (Fig. 5). Additionally, the EA phenotypic variations could be explained by each QTL (R2) ranging from 5.5% (qCIKEA6) to 7.6% (qBYKEA3). The largest-effect QTL, qBYKEA3, with an additive effect of 0.67%, was distributed on chromosome 3 from the BYK RIL population (Table 2). For six QTLs, alleles from the female-parents including DE3, K22 and X26-4, were regarded to have a higher value for EA than those from the male-parents, BY815, CI7 and MO17 (Table 2).

Chromosome-wise logarithm of the odds (LOD) scores of maize EA QTLs in three locations in different RIL populations. A, B, C and D represent the BYD, BYK, CIK and MX RIL populations. Horizontal lines indicate LOD thresholds determined empirically with 1000 random permutations separately with values of 3.0. BJ, LN and NM show the environments in Beijing, Liaoning, and Neimeng, respectively. BLUP represents the results of QTL mapping using the best linear unbiased prediction value of maize EA traits from three locations

Co-localization of maize EA QTLs on maize chromosomes identified in current study
For further confirmation, the nine QTLs were also individually mapped following growth in different environments to detect the stability without the effect of the environments (Fig. 4). It can be noted that association with EA was stable for the QTLs from the four RIL populations planted in all three environments. Although the LOD values of some QTLs were lower than the threshold, these QTLs still showed obvious LOD peaks in the RILs when grown in different environments (Fig. 4). In addition, qBYDEA2 from YPD and qCIKEA6 from MX showed a small difference in Neimeng compared with the other two environments; qBYKEA3 from BYK was a slightly different in Liaoning in contrast to the other two places. Moreover, the additive × additive epistatic interactions for the identified QTLs in the four RIL populations were also investigated by MIM. The result revealed that no epistatic interaction in each maize EA QTL was observed (data not shown), which indicated that the EA genetic components of these four RIL populations were mainly characterized by additive gene actions.
Identification of candidate genes for maize EA QTLs
Combined with the bin map, the intervals containing identified QTLs for maize EA were narrowed to single bins for each QTL peak using co-segregating markers (Fig. 6). The physical distances of the top bins ranged from 9.24 Mb (qBYKEA3) to 240.2 Mb (qMXEA4). These bins each contained various annotated genes from the B73 reference genome v.5b.60 (http://ensembl.gramene.org/Zea_mays/Info/Index). A significant difference was found in gene types between QTLs for maize EA in the four RIL populations. Fourteen genes have been functionally annotated from a total of 331 genes which were regarded as the candidates for maize EA QTLs, including GRMZM2G350165, GRMZM5G881387, GRMZM2G036340, GRMZM2G176063, GRMZM2G133331, GRMZM2G351330, GRMZM2G178102, GRMZM2G418415, AC199526.5_FG003, GRMZM2G332280, GRMZM5G834377, GRMZM2G044752, GRMZM2G106053 and GRMZM2G324285. According the function analysis, these candidate genes could be roughly divided into three categories: signal transduction (3 genes), gene expression regulation including transcriptional, epigenetic and translational regulation (7 genes), and metabolism related activities (3 genes). Additionally, one gene was of unknown function (Table 3). The ratios of different gene types are exhibited in Fig. 7.

Chromosome-wise logarithm of the odds (LOD) profiles for maize EA QTL bins, and representations of genes spanning the peak bins. The candidate genes to have putative functions associated with maize are indicated by red bands; other genes in the peak bin are marked by dark bands

Relative proportion of the 14 candidate genes for maize EA QTLs from the four maize RIL populations
Discussions
Dissecting the genetic architecture of the maize EA trait
In the current study, 56 QTLs related to maize EA were identified from four RIL populations, among which nine QTLs were noted to have larger effects by BLUP value detection. These nine QTLs were located on chromosomes 2, 3, 4, 6 and 7 with each accounting for 5.5–7.6% of the EA phenotypic variation (Table 2; Fig. 5). All these nine QTLs were stable across different environments, which was consistent with heritability of maize EA in the four RIL populations (Table 1; Fig. 4). However, three QTLs were a bit different in one place from the other two, the reason for that perhaps caused by the interaction between QTLs and environments. Additionally, no epistatic interaction between EA QTLs was observed. Basically, we did not find any prominent large-effect QTLs in this study, as the top two effective QTLs, qBYKEA3 and qMXEA2-2, only explained 7.6% and 6.9% of the EA variation, respectively. Nevertheless, the nine QTLs related to maize EA identified in this paper are all novel. In addition, these QTLs contributed 12.0%, 7.6%, 17.7% and 20.2% to the phenotypic variations for maize EA in the BYD, BYK, CIK and MX RIL populations, respectively (Table 2). All the results above indicate that the genetic component of maize EA is quite complicated, reflecting the complexity of maize EA development and biosynthesis. Moreover, not only the 9 large-effect QTLs but also the other 47 QTLs with smaller effects might make important contributions to maize EA trait, which deserve further study. Moreover, three QTLs, qCIKEA3-1, qCIKEA3-2 and qCIKEA6, all from the CIK RIL population, were found to locate in excessively large bins which covered the centromeres in chromosomes 3 and 6 (Fig. 5). As we know, few recombination events occur at the centromeres and surrounding regions during meiosis, and it is difficult for fine mapping. Hence, further study was unexpected to narrow the bins in which these QTLs located. Interestingly, qCIKEA3-1 and qCIKEA3-2 were partially overlapped at the mapping level. Anyway, it was difficult to determine whether they were the same QTLs after fine mapping, which also needs further investigation.
Interaction between identified QTLs for maize EA
Annotated gene types were apparently different between the nine effective QTLs for maize EA in the four RIL populations (Table S1). Based on the gene composition, the nine QTLs could be divided into five groups or classes.
The first group has two QTLs, qBYKEA3 and qCIKEA3-1, and only contained two genes for signal transduction, without any known gene for gene expression regulation or metabolism. Hence, the role of this QTL might be related to cell signaling of other loci.
The unique QTL, qCIKEA3-2, in the second group contained three genes for signal transduction and gene expression regulation but no genes for metabolism-related activities, implying its role in cell signal transducing and gene expression regulating.
As for the third group, qMXEA2-2 and qMXEA4, both from the MX RIL population, contained three genes related to metabolism activities but no genes for signal transduction or gene expression regulation, which could probably depend on the input of cell signals and transcriptional regulation from other loci.
In the fourth group, three QTLs, qBYDEA2, qCIKEA6 and qMXEA2-1, contained a total of five genes for gene expression and transcriptional regulation, without any known genes for signal transduction or metabolism-related activities, which suggests that these QTLs might play a role in regulating the gene expression of other loci.
The last QTL contained a gene with unknown function, which is homologous to a gene encoding vacuole sorting receptor-1 from Arabidopsis.
Taken together, the results above demonstrated that interactions between QTLs probably occurred for maize EA in different populations, although we have not observed any epistatic effect among them. Further study and deeper investigation are needed to solve the contradiction between theory and observation.
Association of candidate genes with maize EA QTLs
As we know, the population and marker density mainly determine the recombination frequency of a population which subsequently affect the QTL mapping resolution (Hennen-Bierwagen and Myers 2013). Thus, improving the marker density enables the revealing of the recombination events, the increase the genetic map resolution and the enhancing of the precision of QTL mapping (Huang et al. 2014). The high-density SNP bin map is a successful example used in QTL mapping, which has been proven in previous studies on multiple maize traits (Xie et al. 2010; Yu et al. 2011; Zou et al. 2012; Guo et al. 2014; Chen et al. 2014; Wen et al. 2015). In this study, the sizes of the QTL interval were finely reduced in spite of three larger ones. And the single bins at each QTL peak exhibited more associations than those outside the peak. In addition, the accuracy of the associated bins was further confirmed by the stability of the QTL peak positions across the four RILs grown in different environments (Fig. 2). Hence, the relatively small distance allowed us to identify candidate genes for the observed maize EA QTLs. Finally, 14 leading candidate genes were annotated for the nine EA QTLs, which could be mainly classified into three groups (Table 3).
Candidate genes for signal transduction
Three genes for signal transduction located in qBYKEA3, qCIKEA3-1 and qCIKEA3-2 for EA were from the BYK and CIK RIL populations. The first candidate gene encodes CAF2–CRS2-associated factor 2, which promotes the splicing of group II introns in maize chloroplasts (Ostheimer et al. 2006). The second gene encodes the Leucine-rich repeat transmembrane protein kinase (LRR-RK), which plays an essential role in hormone signal transduction and plant development in maize (Mitchell et al. 1970). The third candidate is identified from a maize mutant d1-dwarf plant1, which is homologous to GA3OX1 (Gibberellin 3-oxidase-1) participating in the GA biosynthesis (Ren et al. 2016).
Candidate genes for gene expression regulation
As many as seven potential genes for gene expression and transcription regulation in qBYDEA2, qCIKEA3-2, qCIKEA6 and qMXEA2-1 were from the BYK, CIK and MX populations. These genes encode various transcription factors regulating the growth and development, protein synthesis and differentiation, etc. For example, Fea4 (fasciated ear4) encodes a bZIP transcription factor that regulates shoot meristem size in maize (Pautler et al. 2015). Another gene encodes a transcription factor for bHLH88 (basic helix–loop–helix), which is proved to participate in the photo-morphogenesis in maize (Teppeman et al. 2006).
Candidate genes for metabolism
Three genes associated with metabolism-related activities were located in qMXEA2-2 and qMXEA4, both from MX RIL population. Two genes encode α/β-hydrolases and another one encodes anthranilate phosphoribosyl-transferase, regulating the polysaccharide metabolism (Koch 2004; Kramer and Ackelsberg 2015).
Future potential application of EA QTLs in maize breeding
In the current study, nine effective QTLs associated to maize EA were identified from four RIL populations using a high-density linkage map and bin mapping. Two major effective QTLs, qBYKEA3 on chromosome 3 and qMXEA2-2 on chromosome 2, explained 7.6% and 6.9% of EA variation in BYK and MX populations, respectively, which were stable across different environments. They are both promising for the introgression of their favorable alleles to improve the maize EA trait using the marker-assisted selection, whereas the favorable alleles of these two loci associated with EA came from different parents. Therefore, in order to improve the EA into one genotype, it is better to pyramid them in this genotype from different genotypes.
References
Basten CJ, Weir BS, Zeng ZB, Typeset IL (2005) Windows QTL cartographer v.2.5
Blösch R, Plaza WS, Barbier RP et al (2020) Panicle angle is an important factor in Tef lodging tolerance. Front Plant Sci. https://doi.org/10.3389/fpls.2020.00061.eCollection
Chen GX, Liu JB, Hong DL (2006) Genetic analysis on panicle angle and number of spikelets per panicle by using six generations of three crosses derived from erect × curve panicles in japonica rice (Oryza sativa L.). ACTA Agron Sin 32(2):1143–1150 (in Chinese with English abstract)
Chen ZL, Wang BB, Dong XM et al (2014) An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genom 15:433. https://doi.org/10.1186/1471-2164-15-433
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
de Givry S, Bouchez M, Chabrier P et al (2005) CARHT A GENE: multipopulation integrated genetic and randiation hybrid mapping. Bioinformatics 21:1703–1704
Ding J, Zhang L, Chen J et al (2015) Genomic dissection of leaf angle in maize (Zea mays L.) using a four-way cross mapping population. PLoS ONE 10(10):e0141619. https://doi.org/10.1371/journal.pone.0141619
Falconer D, Mackay T (1996) Introduction to quantitative genetics. Longman, Burnt Mill
Ganal MW, Durstewitz G, Polley A et al (2011) A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 6(12):e28334. https://doi.org/10.1371/journal.pone.0028334
Goldman IL, Rocheford TR, Dudley JW (1993) Quantitative loci influencing protein and starch concentration in the Illinois long term selection maize strains. Theor Appl Genet 87(1–2):217–224
Guo J, Su G, Zhang J et al (2008) Genetic analysis and QTL mapping of maize yield and associate agronomic traits under semi-arid land condition. Afr J Biotechnol 7(12):1829–1838
Guo TT, Yang N, Tong H et al (2014) Genetic basis of grain yield heterosis in an “immortalized F2” maize population. Theor Appl Genet 127(10):2149–2158
Guo Z, Zou C, Liu X et al (2020) Complex genetic system involved in fusarium ear rot resistance in maize as revealed by GWAS, bulked sample analysis, and genomic prediction. Plant Dis. https://doi.org/10.1094/pdis-07-19-1552-re
He J, Li J, Huang Z et al (2015) Composite interval mapping based on lattice design for error control may increase power of quantitative trait locus detection. PLoS ONE 10:e0130125. https://doi.org/10.1371/journal.pone.0130125
Hennen-Bierwagen TA, Myers MA (2013) Seed genomics. Springer, New York
Huang BQ, Hennen-Bierwagen TA, Myers AM et al (2014) Functions of multiple genes encoding ADP-glucose pyrophosphorylase subunits in maize endosperm, embryo, and leaf. Plant Physiol 164(2):596–611
Huang C, Chen QY, Xu GH et al (2016) Identification and fine mapping of quantitative trait loci for the number of vascular bundle in maize stem. J Integr Plant Biol 58(1):81–90
James M, Myers A (2009) Handbook of maize: its biology. Springer, New York
Jiang JH, Guo Y, Chen XG et al (2007) The correlation between panicle angle and rice quality and genetic analysis on rice quality in japonica rice (Oryza sativa L.). Yi Chuan 29(6):714–724
Kao CH, Zeng ZB, Teasdale RD (1999) Multiple interval mapping for quantitative trait loci. Genetics 152:1203–1216
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence-intervals for heritability on a progeny mean sasis. Crop Sci 25:192–194
Koch K (2004) Sucrose metabolism: regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr Opin Plant Biol 7(3):235–246
Kramer EM, Ackelsberg EM (2015) Auxin metabolism rates and implications for plant development. Front Plant Sci 6:150
Ku LX, Zhao WM, Zhang J et al (2010) Quantitative trait loci mapping of leaf angle and leaf orientation value in maize (Zea mays L.). Theor Appl Genet 121(5):951–959
Lander ES, Botstein D (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121(1):185–199
Li X, Cheng MJ, Wang Y et al (2010) Study on maize yield and its ear characters under water-limited conditions. J China Agric Univ 15(4):8–12 (in Chinese with English abstract)
Li Q, Yang X, Xu S et al (2012) Genome-wide association studies identified three independent polymorphisms associated with alpha-tocopherol content in maize kernels. PLoS ONE 7(5):e36807. https://doi.org/10.1371/journal.pone.0036807
Li C, Li Y, Shi Y et al (2015) Genetic control of the leaf angle and leaf orientation value as revealed by ultra-high density maps in three connected maize populations. PLoS ONE 10(3):e0121624. https://doi.org/10.1371/journal.pone.0121624
Li PC, Zhuang ZJ, Cai HG et al (2016a) Use of genotype-environment interactions to elucidate the pattern of maize root plasticity to nitrogen deficiency. J Integr Plant Biol 58(3):242–253
Li XN, Chen JX, Su WY et al (2016b) Correlation and path analysis of ear characters and yield in different maize combinations. Bull Agric Sci Technol 4:37–40 (in Chinese with English abstract)
Li Z, Liu P, Zhang X et al (2020) Genome-wide association studies and QTL mapping uncover the genetic architecture of ear tip-barrenness in maize. Physiol Plant. https://doi.org/10.1111/ppl.13087
Liu JB, Hong DL (2005) Genetic analysis on panicle angle and number of spikelets per panicle in japonica rice (Oryza sativa). Chin J Rice Sci 19(3):223–230. https://doi.org/10.16819/j.1001-7216.2005.03.005(in Chinese with English abstract)
Mendes-Moreira P, Alves ML, Satovic Z et al (2015) Genetic architecture of ear fasciation in maize (Zea mays) under QTL scrutiny. PLoS ONE 10(4):e0124543. https://doi.org/10.1371/journal.pone.0124543
Mickelson SM, Stuber CS, Senior L et al (2002) Quantitative trait loci controlling leaf and tassel traits in a B73 × Mo17 population of maize. Crop Sci 42:1902–1909
Mitchell JW, Mandava N, Worley JF et al (1970) Brassins; a new family of plant hormonca from rape pollen. Nature 225(5237):1065–1068
Niu FA, Liu J, Guo Y et al (2013) New stably expressed loci responsible for panicle angle trait in Japonica rice in four environments. Rice Sci 20(2):111–119
Orawan J, Fu XY, Xu J et al (2017) Genetic dissection of carotenoids in maize kernels using high-density single nucleotide polymorphism markers in a recombinant inbred line population. Crop J 5(1):63–72
Ostheimer GJ, Rojas M, Hadjivassiliou H, Barkan A (2006) Formation of the CRS2-CAF2 group II intron splicing complex is mediated by a 22-amino acid motif in the COOH-terminal region of CAF2. J Biol Chem 281:4732–4738
Pan Q, Li L, Yang X et al (2016) Genome-wide recombination dynamics are associated with phenotypic variation in maize. New Phytol 210(3):1083–1094
Pautler M, Eveland AL, LaRue T et al (2015) FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell 27(1):104–120
Ren D, Rao Y, Wu L et al (2016) The pleiotropic ABNORMAL FlOWER AND DWARF1 affects plant height, floral development and grain yield in rice. J Integr Plant Biol 58:529–539
Robinson HF, Comstock RE, Harvey PH (1951) Genotupic and phenotypic correlations in corn and their implications in selection. Agron J 43(34):282–287
Song JJ, Liu JL, Zhou BM et al (2006) Study on the relationship between ear characters and yield in Maize. J Jilin Agric Sci 31(4):11–13 (in Chinese with English abstract)
Song WB, Wang BB, Hauck AL et al (2016) Genetic dissection of maize seedling root system architecture traits using an ultra-high density bin-map and a recombinant inbred line population. J Integr Plant Biol 58(3):266–279
Sun SC (2019) Effect of plant density towards maize yield and traits in Shandong province. Agric Eng Technol. https://doi.org/10.16815/j.cnki.11-5436/s.2019.11.013(in Chinese with English abstract)
Tai HH, Xin L, Nina O et al (2016) Transcriptomic and anatomical complexity of primary, seminal, and crown roots highlight root type-specific functional diversity in maize (Zea mays L.). J Exp Bot 67(4):1123–1135
Tang BJ, Ding Y (2008) Impact of plant density on maize yield and main agronomic traits. Chin Seed Ind 10:35–37. https://doi.org/10.19462/j.cnki.1671-895x.2008.10.014(in Chinese with English abstract)
Tang J, Yan J, Ma X et al (2010) Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL mapping in an immortalized F2 population. Theor Appl Genet 120(2):333–340
Teppeman JM, Hwang YS, Quail PH (2006) HhyA dominates in transduction of red-light signals to rapidly-responding genes at the initiation of A rabidops is seeding de-etiolation. Plant J 48:728–742
Tong H (2015) Linkage analysis of ear characters in Maize. Dissertation, Huazhong Agricultural University
Van OH, Stam P, Visser RG, Van Eck HJ (2015) RECORD: a novel method for ordering loci on a genetic linkage map. Theor Appl Genet 112(1):30–40
Veldboom LR, Lee M (1994) Molecular-marker-facilitated studies of morphological traits in maize: II Determination of QTLs for grain yield and yield components. Theor Appl Genet 89(23):451–458
Wang T, Min W, Hu S et al (2015) Genetic basis of maize kernel starch content revealed by high-density single nucleotide polymorphism markers in a recombinant inbred line population. BMC Plant Biol 15(1):1–12
Wang B, Zhang ZH, Fu ZY et al (2016) Comparative QTL analysis of maize seed artificial aging between an immortalized F2 population and its corresponding RILs. Crop J 4(1):30–39
Wen WW, Li K, Alseekh S et al (2015) Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell 27(7):1839–1856
Xiao Y, Tong H, Yang X et al (2016) Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol 210(3):1095–1106
Xie WB, Feng Q, Yu HH et al (2010) Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci USA 107(23):10578–10583
Yang X, Yan J, Shan T et al (2010) Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor Appl Genet 121(3):417–431
Yu HH, Xie WB, Wang J et al (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 6(3):e17595. https://doi.org/10.1371/journal.pone.0017595
Zhang LG, Fan QJ, Chen XC et al (2012) Correlation analyisis between kernel dry-down rate and main agronomic traits after maize physiological mature. J Heilongjiang Agric Sci 3:1–5 (in Chinese with English abstract)
Zhang J, Ku LX, Han ZP et al (2014) The ZmCLA4 gene in the qLA4-1 QTL controls leaf angle in maize (Zea mays L.). Exp Bot 65(17):5063–5076
Zou GH, Zhai GW, Feng Q et al (2012) Identification of QTLs for eight agronomically important traits using ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. Exp Bot 63(15):5451–5462
Acknowledgements
We are very grateful to Professor Yang (China Agricultural University) for providing the seeds and genotypes of the four maize RIL populations. This work was supported by grants from the Technology Pillar Program of Liaoning Province, China (2015103001) and General Program of National Nature Science Foundation of China (31771880), and a grant from the Cultivation Plan for Youth Agricultural Science and Technology Innovative Talents of Liaoning Province (2015043).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lin, J., Li, S., Liang, G. et al. Genetic basis of maize ear angle revealed by high-density single nucleotide polymorphism markers in four recombinant inbred line populations. Euphytica 216, 132 (2020). https://doi.org/10.1007/s10681-020-02662-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-020-02662-2