
Abstract
In this study, we developed a total of 37 simple sequence repeat (SSR) markers from 11 bacterial artificial chromosome (BAC) clone sequences anchored on chromosome 12 of tomato available at Solanaceae Genomics Network. These SSR markers could group a set of 16 tomato genotypes comprising of Solanum lycopersicum, S. pimpinellifolium, S. habrochaites, and S. pennellii unambiguously according to their known species status. Clear subgroups of genotypes within S. lycopersicum were also observed. A subset of 16 SSR markers representing the 11 BAC clones was used for developing genetic linkage maps of three interspecific F2 populations produced from the crosses involving a common S. lycopersicum parent (CLN2498E) with S. pennellii (LA1940), S. habrochaites (LA407) and S. pimpinellifolium (LA1579). The length of the genetic linkage maps were 112.5 cM, 109.3 cM and 114.1 cM, respectively. Finally, an integrated genetic linkage map spanning a total length of 118.7 cM was developed. The reported SSR markers are uniformly distributed on chromosome 12 and would be useful for genetic diversity and mapping studies in tomato.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tomato (Solanum lycopersicum) is one of the most important vegetable crops globally and contributes significantly to human nutrition. The genetic diversity present in wild relatives of the Solanum lycopersicum complex (S. lycopersicum var. cerasiforme, S. pimpinellifolium, S. habrochaites, S. pennellii, S. chmeilewskii, S. neorickii, and S. cheesmaniae) and the Solanum peruvianum complex (S. chilense, S. peruvianum) has been exploited extensively in genetic improvement of cultivated tomato (Rick et al. 1987). Advancements in molecular marker technology have enhanced the efficiency for trait introgression from wild species.
A large number of DNA markers have been developed and are routinely used in molecular genetic analyses in tomato. Several molecular marker based genetic linkage maps have been constructed using mapping populations derived from interspecific crosses between cultivated tomato and wild relatives (Tanksley et al. 1992; Chen and Foolad, 1999; Zhang et al. 2002; Sharma et al. 2008). Most of these maps were constructed based on restriction fragment length polymorphism (RFLP) markers available in the high density linkage map developed by Tanksley et al. (1992). However, microsatellites or simple sequence repeats (SSRs) are more preferred markers because of their high polymorphism and simplicity (Tautz and Renz 1984).
In tomato, considerable effort has been made to develop and map SSRs. Conventionally, SSR markers were developed by genomic library screening approach, which is tedious and expensive (Broun and Tanksley 1996; Areshchenkova and Ganal 1999, 2002; Suliman-Pollatschek et al. 2002). A more rapid and economical way to develop SSR markers is to make use of genomic sequences and expressed sequence tags (ESTs) available in genome databases such as European Molecular Biology Laboratory (EMBL), GenBank, and Solanaceae Genomics Network (SGN) (Arens et al. 1995; Smulders et al. 1997; He et al. 2003; Frary et al. 2005). Recently, Ohyama et al. (2009) and Shirasawa et al. (2010) developed a large number of SSR markers using bacterial artificial chromosome (BAC)-ends, cDNA and EST sequences from genome databases. Geethanjali et al. (2010) developed a total of 54 SSR markers from 17 complete BAC clone sequences on chromosome 6 and mapped 21 of them. Currently, a total of 1707 tomato SSR markers have been mapped; 1686 of them in high density linkage map derived from S. lycopersicum × S. pennellii cross and 21 of them in the linkage map derived from West Virginia 700 (S. pimpinellifolium) and Hawaii 7996 (S. lycopersicum) cross. Information for these SSR markers is available at SOL Genomics Network (http://www.sgn.cornell.edu), Vegmarks (http://vegmarks.nivot.affrc.go.jp/) and http://www.kazusa.or.jp/tomato/, respectively.
Highly saturated linkage maps with uniformly distributed markers are essential to identify quantitative trait loci (QTLs) associated with horticultural traits and to understand the genetic structure and diversity of germplasm resources. The currently available linkage maps in tomato show that a large proportion of SSR markers developed either from genomic libraries or BAC-end sequences were located in heterochromatin regions and clustered around the centromeres (Areshchenkova and Ganal 2002; Frary et al. 2005; Ohyama et al. 2009; Shirasawa et al. 2010). Furthermore, saturation of several intra- and interspecific linkage maps has been difficult due to the low level of polymorphism within cultivated tomato and between its closely related wild species. Randomly distributed cDNA/EST derived SSRs are much less polymorphic among cultivars than BAC-end derived SSRs (Ohyama et al. 2009). Hence, development of more SSR markers in genomic regions lacking adequate markers could complement the existing marker resources and will be useful for genetic mapping in multiple mapping populations. Identifying common polymorphic markers across multiple mapping populations enables transfer of linkage information among maps; the markers could serve as anchors to integrate maps for development of highly saturated consensus maps (Radhika et al. 2007). For this purpose, BAC sequences anchored on the genetic and physical maps are more useful for SSR marker development. The availability of such anchored BAC clones is increasing as the international tomato genome sequencing project progresses (Mueller et al. 2009).
In this study, our objectives were to develop uniformly distributed SSR markers from BAC clones anchored on chromosome 12 of tomato and assess their usefulness for genetic diversity analysis and linkage mapping using three different interspecific F2 populations. Development of more uniformly distributed SSR markers on this chromosome would be very useful, because many genes and QTLs associated with economically important traits such as resistance to black mould (Robert et al., 2001), bacterial wilt (Wang et al. 2000), cucumber mosaic virus (Stamova and Chetelat 2000), early blight (Foolad et al. 2002), powdery mildew (Chungwongse et al. 1994; Bai et al. 2003) and root knot nematode (Yaghoobi et al. 2005) have been mapped on chromosome 12.
Materials and methods
Plant materials and DNA extraction
A panel of 16 genotypes comprising of cultivated tomato and its wild relatives were used for genetic diversity analysis. The genotypes included were: S. lycopersicum—CLN2498E, Arka Meghali, CA4, CLN1621L, FLA456, CL5915-93D4, Hawaii 7996, M-82, T4065; S. pimpinellifolium—West Virginia 700, LA1579, L3708; S. habrochaites—LA407, LA1033; S. pennellii—LA716, LA1940. These genotypes represented diverse origins and possessed useful traits such as resistance to biotic and abiotic stresses. More details of the accessions are available in Geethanjali et al. (2010). The seeds of these genotypes were obtained from Genetic Resources and Seed Unit, AVRDC—The World Vegetable Center (AVRDC), Taiwan. For genetic mapping, three interspecific F2 populations were developed by crossing a common S. lycopersicum parent (CLN2498E) with S. pimpinellifolium (LA1579), S. habrochaites (LA407), and S. pennellii (LA1940). A subset of 94 F2 plants from each cross was used for SSR genotyping. Genomic DNAs were isolated from the fresh leaf tissues using the protocol of Fulton et al. (1995).
Database search for SSR motifs and primer designing
Eleven BAC clones (S. lycopersicum) distributed uniformly on chromosome 12 of EXPEN2000 genetic map (http://www.sgn.cornell.edu) were searched for perfect di-, tri- and tetranucleotide microsatellite motifs using the simple sequence repeat identification tool (SSRIT) program (http://www.gramene.org/db/markers/ssrtool). A length criterion of a minimum of eight repeat motifs was adopted. Forty-one primer pairs were designed from sequences flanking the repeat motifs using PRIMER3.0 (http://www.-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi). The parameters of product size 100–300 bp, primer length of 20–24 bp, and melting temperature of 54–64°C and a GC content of 40–60% were set to design these primers. The primers were synthesized from Genomics BioSci & Tech Company, Taiwan (www.genedragon.com.tw). The details of BAC clones, SSR repeat motifs, and primers designed are listed in Table 1.
PCR amplification of microsatellites
PCR amplifications of SSRs were performed in 15 μl reactions containing 0.2 μM of each primer, 200 μM of deoxyribonucleotides, 50 mM KCl, 10 mM Tris HCl (pH 8.3), 1.5 mM MgCl2, 25 ng of DNA and 0.5 unit of Taq DNA polymerase. The amplifications were conducted in a PTC 200 DNA engine thermal cycler (MJ Research, USA). The temperature profile used for PCR amplification included 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 55°C for 45 s, 72°C for 45 s, and finally by 7 min at 72°C for the final extension. Annealing temperature was adjusted based on the specific requirement of each primer pair. PCR products (3 μl) were analyzed on 6% non-denaturing polyacrylamide gel in 0.5× TBE buffer. After electrophoresis, the gels were stained with 5 μg/ml−1 ethidium bromide and the bands were visualized under UV light using the Alpha Imager 3300 system.
Estimation of polymorphic information content and genetic diversity analysis
Individual SSR alleles observed for the 16 tomato genotypes were scored as different characters and a pair-wise dissimilarity matrix was generated. An allele at a microsatellite locus that consistently failed to amplify to detected levels via the polymerase chain reaction was considered as a null allele (Dakin and Avise 2004). The genetic relatedness among genotypes was analyzed using Jaccard’s similarity coefficient and tree construction through neighbor-joining cluster analysis based on the principle of parsimony as implemented in DARwin (Dissimilarity Analysis and Representation for windows) V.5.0.158 (Perrier and Jacquemoud-Collet 2006). To determine the statistical support for different branch points, the percentage of bootstrap values were estimated based on 1000 re-samplings of the data. The polymorphism information content (PIC) value of each SSR marker was calculated based on the formula, Hn = 1 − Σpi 2, where pi is the allele frequency for the ith allele (Nei 1973).
Linkage map construction
The genotypic data of a subset of 94 F2 plants produced from the crosses of CLN2498E × LA1579, CLN2498E × LA407, and CLN2498E × LA1940 were used to map the newly developed SSRs and compare their map distances. The linkage map was constructed using JoinMap® version 3.0 (van Ooijen and Voorrips 2001). All the loci were tested for significant deviation from the expected Mendelian genotypic ratios using chi-square (χ2) goodness-of-fit analysis. The logarithm of odds ratio (LOD) scores and pair-wise recombination frequencies between markers were calculated. A LOD of 6.0 was used to create the linkage groups in the three populations. Recombination values were converted to genetic distances using the Kosambi mapping function (Kosambi 1944). The Combine Map command was used to check the orientation and order of markers between the three linkage maps. An integrated map was built from these three individual maps using the Combine Groups for Map Integration function. The pairwise recombination frequencies between the common markers and LOD scores were calculated using the Heterogeneity Test command. The individual and integrated linkage maps were drawn using Mapchart software (Voorrips 2002). Map positions of common markers across the three individual interspecific maps were compared using the Homolog 1 and Homolog 2 functions.
Results
Occurrence of microsatellite motifs in BAC sequences
A total of 88 perfect di- and trinucleotide microsatellite motifs were identified from 11 BAC clones. The number of repeat motifs per BAC clone ranged from 4 to 15. Dinucleotide repeat motifs were predominant followed by trinucleotide repeats. AT repeats were the most common among dinucleotide motifs followed by GA/CT repeats. There was no predominant trinucleotide motifs observed in this study. Out of 88 repeat motifs identified, a total of 41 were selected for marker development. The primer pairs were screened to check for amplification and four failed to produce any amplification product. The remaining 37 primer pairs produced clear banding patterns of DNA fragments with expected size; 26 of them were class I types (>10 repeats) and 11 of them were class II types (≤10 repeats).
Polymorphism of SSR markers
In total, the 37 SSRs produced 183 alleles in the panel of 16 tomato accessions including cultivated tomato varieties and wild accessions. The allelic distribution for the SSR loci SLM12-31 across 16 genotypes is shown in Fig. 1. Number of alleles across 16 accessions ranged from 1 (SLM12-7, SLM12-35) to 10 (SLM12-31). Null allele was a common feature in S. pennellii (29.2%) and S. habrochaites (32.3%) accessions. The PIC values reflected allele diversity and frequency among different accessions and ranged from zero to 0.88 (Table 1). Among the 37 polymorphic SSRs, 20 primers showed polymorphism within S. lycopersicum genotypes with the number of alleles ranging from 2 to 5 and PIC values ranging from 0.19 to 0.75. SLM12-31 alone was sufficient to differentiate five out of eight S. lycopersicum genotypes. The number of polymorphic SSR alleles between CLN2498E (S. lycopersicum) and other cultivated and wild tomato genotypes ranged from 4 to 32 and the percentage of polymorphism ranged from 11 to 86 (Table 2).

Allelic distribution of SLM12-31 across 16 tomato genotypes. M 25-bp ladder markers; Lane 1 LA1579 (S. pimpinellifolium); 2 LA1940 (S. pennellii); 3 CLN2498E (S. lycopersicum); 4 Arka Meghali (S. lycopersicum); 5 CA4 (S. lycopersicum); 6 CLN1621L (S. lycopersicum); 7 FLA456 (S. lycopersicum); 8 CL5915-93D4 (S. lycopersicum); 9 Hawaii 7996 (S. lycopersicum); 10 LA716 (S. pennellii); 11 West Virginia700 (S. pimpinellifolium); 12 M82 (S. lycopersicum); 13 LA1033 (S. habrochaites); 14 LA3708 (S. pimpinellifolium); 15 LA407 (S. habrochaites); 16 T4065 (S. lycopersicum)
A comparison was made between repeat length and the degree of polymorphism. Most of the microsatellite loci were polymorphic between accessions of S. lycopersicum and wild relatives irrespective of the length of repeat motif. However, the degree of polymorphism increased with the total length of the repeat within the cultivars of S. lycopersicum. The average PIC values of class I and II type SSRs were 0.64 and 0.38, respectively.
Genetic diversity analysis
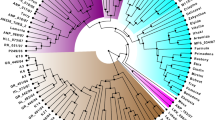
Based on 37 SSR markers specific to chromosome 12, the 16 tomato genotypes were grouped into four major clusters (Fig. 2). The wild genotypes were well separated from the cultivated species, and three distinct groups were observed, i.e. S. pennellii (LA716 and LA1940), S. habrochaites (LA407 and LA1033) and S. pimpinellifolium (LA3708, LA1579). West Virginia 700 (S. pimpinellifolium) grouped separately but more closely related to S. pimpinellifolium genotypes. The cultivated genotypes were grouped into two sub-clusters. However, the bootstrap values were much lower at the nodes, except two, in these two clusters. This indicated the relatively higher genetic similarity among the cultivated accessions.

The topology of the UPGMA dendrogram for individual tomato accessions based on 37 SSR markers. Numbers at nodes indicate bootstrap values (percentage of 1000 bootstrap re-sampling)
Genetic mapping of chromosome 12
The SSR markers developed in the present study were polymorphic in one or more of the three mapping populations. Parental screening identified 32 polymorphic loci between CLN2498E and LA1940, 32 between CLN2498E and LA407, and 21 between CLN2498E and LA1579. SLM12-37 developed from the BAC clone C12HBa0021L02 was found to be polymorphic only between CLN2498E and LA1579. A subset of 16 polymorphic markers with one to two markers representing each of the 11 BAC clones was selected for genotyping. All 16 markers were not commonly polymorphic across three mapping populations. The number of polymorphic markers was 13, 12 and 14 for S. pennellii, S. habrochaites and S. pimpinellifolium populations, respectively. Those SSR loci producing null alleles in LA1940 and LA407 were scored as dominant markers. The rest of the markers were scored as co-dominant markers.
The segregation of the markers significantly deviated from Mendelian ratios (3:1 for dominant markers or 1:2:1 for co-dominant markers) in all three populations. The extent of skewed segregation was higher in S. lycopersicum × S. pennellii cross. Out of 13 marker loci surveyed, eight exhibited significant deviation from the expected segregation ratios. In S. lycopersicum × S. habrochaites cross, four out of 12 markers exhibited skewness, while in S. lycopersicum × S. pimpinellifolium cross, only one out of 14 markers exhibited skewness.
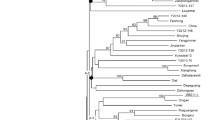
The total length of linkage map of chromosome 12 was 112.5 cM, 109.3 cM, and 114.1 cM for the S. lycopersicum × S. pennellii, S. lycopersicum × S. habrochaites, and S. lycopersicum × S. pimpinellifolium populations, respectively (Fig. 3). The interval distance between markers developed from adjacent BAC clones in CLN2498E × LA1940 (S. pennellii) map were similar to the map distance between the actual BAC clones anchored on the tomato-EXPEN2000 map in SGN. However, contraction of marker intervals (between SLM12-41 and SLM12-38) near the centromeric region was observed in CLN2498E × LA407 (S. habrochaites) map. These markers were derived from three BAC clones viz., C12HBa0009J11, C12HBa0165B12, and C12HBa0193C03. In CLN2498E × LA1579 (S. pimpinellifolium) map, a contraction between the SLM12-20 and SLM12-17 as well as an expansion between SLM12-36 and SLM12-24 were observed. The differences in the distance between marker intervals contributed to the slight changes in the total length of linkage map of chromosome 12 between the three populations.

SSR linkage maps of chromosome 12 for the inter-specific crosses in tomato. A CLN2498E × LA1940 (S. lycopersicum × S. pennellii); B CLN2498E × LA407 (S. lycopersicum × S. habrochaites); C CLN2498E × LA1579 (S. lycopersicum × S. pimpinellifolium). The arrow mark indicates the SSR markers that are common across linkage maps
An integrated linkage map was built by combining the genotypic data from all three mapping populations (Fig. 4). Because Join Map uses the regression mapping algorithm for map integration, distances between the common markers in all three mapping populations were averaged. Based on the mean recombination frequencies and LOD score, map distance was calculated to obtain the integrated map. The integrated map comprised of 16 markers spanning a total length of 118.7 cM and had two large gaps of 15–20 cM.

Integrated SSR linkage map of chromosome 12 of tomato based on three interspecific mapping populations derived from S. lycopersicum with S. pennelli, S. habrochaites and S. pimpinellifolium crosses
Discussion
With progress in the tomato genome sequencing project, about 50 percent of chromosome 12 has been sequenced and 82 BAC clones are available. Of these, 17 BAC clones have been anchored on the high density genetic map of S. lycopersicum × S. pennellii as on date (http://www.sgn.cornell.edu). Although these anchored BAC clones cannot provide complete coverage of the chromosome, their uniform distribution at an interval of 10–20 cM from each other still serves as a useful resource for marker development. This would particularly complement the high level of clustering in the centromeric region among the available SSR markers of this chromosome.
SSR markers identified in the BAC clones were characterized for the type and number of repeat units. AT motifs accounted for a majority of the dinucleotide repeats. This is consistent with previous studies in tomato (Areshchenkova and Ganal 2002; He et al. 2003; Frary et al. 2005; Ohyama et al. 2009). There was no predominant trinucleotide motifs observed in this study, although ATT, AAT, and CTT motifs were found to be more frequently reported by Smulders et al. (1997), He et al. (2003), and Frary et al. (2005), respectively. SSR marker alleles were detected in almost all the tested accessions of S. lycopersicum and S. pimpinellifolium. However null alleles were a common feature of S. pennellii and S. habrochaites. In a study by Areshchenkova and Ganal (2002) about 50% of SSR markers yielded null alleles in S. pennellii. Such high frequencies of null alleles in distantly related accessions reveals a high amount of sequence diversity in the microsatellite flanking regions, thereby preventing primer binding.
Based on the number of repeat units, SSRs are classified as class I (>10 repeat units) and class II (<10 repeat units) markers (Shultz et al. 2007). Areshchenkova and Ganal (1999, 2002) and Ohyama et al. (2009) developed SSR markers by isolating only class I SSRs from genomic libraries and BAC-end sequences. This may result in a potential loss of polymorphic markers being identified. Hence we developed SSR markers comprising of both class I and class II types with the length of repeat motifs ranging from 16 to 78 nucleotides. Several studies have reported that PIC values are positively influenced by the length of repeat motif of SSR markers in different species (Grist et al. 1993; Thomas and Scott 1993; Minamiyama et al. 2006; Yi et al. 2006). A comparison between repeat length and the degree of polymorphism in this study showed that, irrespective of the length, both class I and class II SSRs were polymorphic between species. Class I SSRs had higher PIC values compared to class II SSRs and were more useful in differentiating genotypes among S. lycopersicum. Similar observations were made by Smulders et al. (1997) and Frary et al. (2005) indicating that longer tomato microsatellites generated polymorphisms within cultivated tomato, while shorter microsatellites could only produce polymorphism between species. However, no relationship between repeat length and PIC also has been reported in pepper (Sanwen et al. 2000) and potato (Milbourne et al. 1998). Although class I SSRs may be preferred for their higher polymorphic values within cultivated genotypes, our results suggest that class II SSRs should be useful to detect species level polymorphisms and to saturate various interspecific linkage maps.
Diversity analysis showed that use of SSR markers from a single chromosome was effective in revealing genetic diversity within both wild and cultivated genotypes of tomato. In this study, West Virginia 700 (S. pimpinellifolium) was grouped separately but more closely related to S. pimpinellifolium. Earlier, Olivier et al. (1995) reported that West Virginia 700 did not considerably differ from S. lycopersicum genotypes based on RFLP polymorphisms. And later studies indicated low polymorphism between West Virginia 700 and Hawaii 7996 (S. lycopersicum) (Thoquet et al. 1996; Wang et al. 2000). Using chromosome 6 specific SSR markers, West Virgina 700 was separated from both S. pimpinellifolium and S. lycopersicum (Geethanjali et al. 2010). There might be a possibility that it could be a derivative of both the species. The varieties, CLN2498E, CLN1621L and CL5915-93D4 bred at AVRDC were grouped together. This was expected, as they possibly could share a close pedigree. For example, these varieties possess moderate to high level of resistance to bacterial wilt (P. Hanson, AVRDC; personal communication) and, a major QTL associated with resistance to bacterial wilt has been mapped on chromosome 12 (Thoquet et al. 1996; Wang et al. 2000).
Segregation distortion is a common phenomenon in interspecific mapping populations, particularly in more distantly related crosses. Our results showed that skewed segregation was more pronounced in the crosses with S. pennellii, followed by S. habrochaites, and was the least in S. pimpinellifolium. These are consistent with previous reports on the extent of skewness in various interspecific crosses. deVicente and Tanksley (1993) observed about 80% skewed segregation in a F2 population derived from S. lycopersicum × S. pennellii cross. Shirasawa et al. (2010) reported segregation distortion for 38.2% of the mapped SSR marker loci in the EXPEN2000 genetic map (S. lycopersicum × S. pennellii); the distortion ratios varied from chromosome to chromosome and in chromosome 12, less than 10% of SSR marker loci showed distortion. Zhang et al. (2002) reported 62% skewed segregation in backcross populations derived from S. lycopersicum × S. habrochaites crosses. The skewed segregation of markers was relatively lower in S. lycopersicum and S. pimpinellifolium crosses (Chen and Foolad 1999). Skewed segregation in interspecific crosses of tomato has been attributed to several factors such as self incompatibility, unilateral incompatibility and gametophytic selection (Trognitz and Schmiediche 1993; Foolad 1996; Bernacchi and Tanksley 1997). Shirasawa et al. (2010) suggested that the bias of segregation distortion can result from the presence of more transmission ratio distortion (TRD) factors (Harushima et al. 2001) on certain chromosomes, and the number of markers linked to those TRD. For example, self-incompatibility locus (SI), which is the major control for unilateral incompatibility in tomato, has been located on chromosome 1 and two additional QTLs enhancing unilateral incompatibility mapped on chromosome 3 and 12 (Bernacchi and Tanksley 1997), and the SSR markers mapped on these chromosomes showed skewed segregation.
Although several SSR based linkage maps are available in tomato, comparison of these maps has not yet been done. This was due to the absence of adequate polymorphic SSR markers segregating in multiple mapping populations. Our strategy to develop as many SSR markers as possible from each BAC clone was aimed to identify more markers that would segregate in multiple mapping populations for the same genomic region. The common SSRs mapped on the three interspecific genetic linkage maps enabled us to compare these maps in terms of marker order, marker interval, and map distance. The marker order among the three maps was in good agreement, and was in the linear order of the BAC clones. The length of linkage maps developed in this study was also comparable with the other published linkage maps. For instance, the total length of linkage map of S. lycopersicum × S. pennelli (LA1940) population was 112.5 cM based on 13 SSR markers in this study. Shirasawa et al. (2010) reported that the total length of linkage map of chromosome 12 of the S. lycopersicum × S. pennelli (LA716) population was about 120.4 cM based on 164 markers. Slight differences were observed in the total length of linkage maps of chromosome 12 between three interspecific populations as a result of contractions and expansions in certain marker intervals. As the mapping populations used in this study had a common S. lycopersicum parent, differences in the total length of linkage maps might have resulted from differences in the recombination rate contributed by the wild species. Furthermore, SSRs derived from three BAC clones (C12HBa0009J11, C12HBa0165B12 and C12HBa0193C03) clustered around the centromere in the CLN2498E × LA407 F2 map, but not in the other two maps. Reduced recombination in the centromeric regions of tomato chromosomes has been observed previously from F2 populations of S. lycopersicum × S. pennellii and S. lycopersicum × S. pimpinellifolium crosses (Frary et al. 1996). Such a centromeric suppression of recombination, if present, could have been accounted for in the EXPEN2000 map. As the positions of BAC clones were inferred based on the EXPEN 2000 map, SSRs derived from these BAC clones also showed similar marker intervals to adjacent BAC clones placed on EXPEN 2000 map without much contraction and expansion in the CLN2498E × LA1940 (S. pennellii) map. The contraction of marker intervals (between SLM12-41 and SLM12-38) near the centromeric region in CLN2498E × LA407 (S. habrochaites) map and a contraction (between the SLM12-20 and SLM12-17) as well as an expansion (between SLM12-36 and SLM12-24) of marker intervals in CLN2498E × LA1579 (S. pimpinellifolium) suggest the cross-specific hotspots of depressed or increased recombination along chromosomes as reported in the earlier studies (Grandillo and Tanksley 1996; Bernacchi and Tanksley 1997). The differences in map distance also could be due in part to the smaller population size, chance recombination, and dominant microsatellite markers affecting mapping accuracy.
As all the markers did not segregate in a single mapping population, we constructed an integrated linkage map of chromosome 12 by combining markers from all three interspecific linkage maps. Integrated linkage maps, once saturated with more markers, would be useful for tagging genes/QTLs and finding markers for marker assisted selection. However, ambiguities in marker order were observed in integrated SSR maps of grapevine (Doligez et al. 2006), soybean (Radhika et al. 2007), and pepper (Lee et al. 2009). Such ambiguities were not observed in the present integrated linkage map of chromosome 12. The marker order in the integrated linkage map was consistent with the individual linkage maps. The total length of integrated linkage map (118.7 cM) was closer to the total length of linkage map of chromosome 12 (120.4 cM) reported by Shirasawa et al. (2010). When more BAC clone sequences become available, development of additional SSR markers and integration with other published SSRs on this chromosome would help to develop highly saturated linkage map.
In conclusion, 37 SSR markers specific to chromosome 12 were developed from anchored BAC clone sequences available in the Solanaceae Genomics Network. These SSR markers add to the present repository of molecular markers available for chromosome 12 in tomato and have potential use in genetic diversity analysis and mapping studies.
References
Arens P, Odinot P, van Heusden AW, Lindhout P, Vosman B (1995) GATA-and GACA-repeats are not evenly distributed throughout the tomato genome. Genome 38:84–90
Areshchenkova T, Ganal MW (1999) Long tomato microsatellites are predominantly associated with centromeric regions. Genome 42:536–544
Areshchenkova T, Ganal MW (2002) Comparative analysis of polymorphism and chromosomal location of tomato microsatellite markers isolated from different sources. Theor Appl Genet 104:229–235
Bai Y, Huang CC, van der Hulst R, Meijer-Dekens F, Bonnema G, Lindhout P (2003) QTLs for tomato powdery mildew resistance (Oidium lycopersici) in Lycopersicon parviflorum G1.1601 co-localize with two qualitative powdery mildew resistance genes. Mol Plant Microbe Interact 16(2):169–176
Bernacchi D, Tanksley SD (1997) An interspecific backcross of Lycopersicon esculentum × L. hirsutum: linkage analysis and a QTL study of sexual compatibility factors and floral traits. Genetics 147:861–877
Broun P, Tanksley D (1996) Characterization and genetic mapping of simple repeat sequences in the tomato genome. Mol Gen Genet 250:39–49
Chen FQ, Foolad MR (1999) A molecular linkage map of tomato based on a cross between Lycopersicon esculentum and L. pimpinellifolium and its comparison with other molecular maps of tomato. Genome 42:94–103
Chungwongse J, Bunn TB, Crossman C, Jiang J, Tanksley D (1994) Chromosomal localization and molecular marker tagging of the powdery mildew resistance gene (Lv) in tomato. Theor Appl Genet 89(1):76–79
Dakin EE, Avise JC (2004) Microsatellite null alleles in parentage analysis. Heredity 93:504–509
deVicente MC, Tanksley SD (1993) QTL analysis of transgressive segregation in an interspecific tomato cross. Genetics 134:585–596
Doligez A, Adam-Blondon AF, Cipriani G, Gaspero DG, Laucou V, Merdinoglu D, Meredith CP, Riaz S, Roux C, This P (2006) An integrated SSR map of grapevine based on five mapping populations. Theor Appl Genet 113:369–382
Foolad MR (1996) Unilateral incompatibility as a major cause of skewed segregation in the cross between Lycopersicon esculentum and L. pennellii. Plant Cell Rep 15:627–633
Foolad MR, Zhang LP, Khan AA, Nino-Liu D, Lin GY (2002) Identification of QTLs for early blight (Alternaria solani) resistance in tomato using backcross populations of a Lycopersicon esculentum × L. hirsutum cross. Theor Appl Genet 104:945–958
Frary A, Presting GG, Tanksley SD (1996) Molecular mapping of the centromeres of tomato chromosomes 7 and 9. Mol Gen Genet 250:295–304
Frary A, Xu Y, Liu J, Mitchell S, Tedeschi E, Tanksley SD (2005) Development of a set of PCR-based anchor markers encompassing the tomato genome and evaluation of their usefulness for genetics and breeding experiments. Theor Appl Genet 111(2):291–312
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Rep 13:207–209
Geethanjali S, Chen KY, Pastrana DV, Wang JF (2010) Development and characterized of tomato SSR markers from genomic sequences of anchored BAC clones on chromosome 6. Euphytica 173:85–97
Grandillo S, Tanksley SD (1996) Genetic analysis of RFLPs, GATA microsatelllites and RAPDs in a cross between L. esculentum and L. pimpinellifolium. Theor Appl Genet 92:957–965
Grist SA, Firgaira FA, Morley AA (1993) Dinucleotide repeat polymorphisms isolated by the polymerase chain reaction. BioTechniques 15:304–309
Harushima Y, Nakagahra M, Yano M, Sasaki T, Kurata N (2001) A genome-wide survey of reproductive barriers in an intraspecific hybrid. Genetics 159:883–892
He C, Poysa V, Yu K (2003) Development and characterization of simple sequence repeat (SSR) markers and their use in determining relationships among Lycopersicon esculentum cultivars. Theor Appl Genet 106:363–373
Kosambi D (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lee HR, Bae IH, Park SW, Kim HJ, Min WK, Han JH, Kim KT, Kim BD (2009) Construction of an Integrated Pepper Map Using RFLP, SSR, CAPS, AFLP, WRKY, rRAMP, and BAC End Sequences. Mol Cells 27:21–37
Milbourne D, Meyer RC, Collins AJ, Ramsay LD, Gebhardt C, Waugh R (1998) Isolation, characterization and mapping of simple sequence repeat loci in potato. Mol Gen Genet 259:233–245
Minamiyama Y, Tsuro M, Hirai M (2006) An SSR-based linkage map of Capsicum annuum. Mol Breed 18:157–169
Mueller LA, Lankhorst RK, Tanksley SD, Giovannoni JJ et al (2009) A snapshot of the emerging tomato genome sequence. Plant Genome 2:78–92
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Ohyama A, Asamizu E, Negoro S, Miyatake K, Yamaguchi H, Tabata S, Fukuoka H (2009) Characterization of tomato SSR markers developed using BAC-end and cDNA sequences from genome database. Mol Breed 23:685–691
Olivier J, Thoquet P, Sperisen C, Rogowsky P, Stephens S, Bonnema G, Lewis C, Nazer R, Tanksley S, Gebhardt C, Grimsley N (1995) Restriction Fragment Length Polymorphisms between tomato lines sensitive or tolerant to bacterial wilt. Rept Tomato Genetics Coop 45:32–33
Perrier X, Jacquemoud-Collet JP (2006) DARwin software http://darwin.cirad.fr/darwin
Radhika P, Gowda SJM, Kadoo NY, Mhase LB, Jamadagni BM, Sainani MN, Chandra S, Gupta VS (2007) Development of an integrated intra-specific map of chickpea (Cicer arietinum L.) using two recombinant inbred line populations. Theor Appl Genet 115:209–216
Rick CM, DeVerna JW, Chetelat RT, Stevens MA (1987) Potential contributions of wide crosses to improvement of processing tomatoes. Acta Hort 200:45–55
Robert VJM, West MAL, Inai S et al (2001) Marker-assisted introgression of blackmold resistance QTL alleles from wild Lycopersicon cheesmanii to cultivated tomato (L. esculentum) and evaluation of QTL phenotypic effects. Mol Breed 8:217–233
Sanwen H, Baoxi Z, Milbourne D, Cardle L, Guimei Y, Jiazhen G (2000) Development of pepper SSR markers from sequence databases. Euphytica 117:163–167
Sharma A, Zhang L, Nino-Liu D, Ashrafi H, Foolad MR (2008) A Solanum lycopersicum × Solanum pimpinellifolium linkage map of tomato displaying genomic locations of R-Genes, RGAs, and candidate resistance/defense-response ESTs. Int J Plant Genomics. doi:10.1155/2008/926090
Shirasawa K, Asamizu E, Fukuoka H, Ohyama A, Sato S, Nakamura Y, Tabata S, Sasamoto S, Wada T, Kishida Y, Tsuruoka H, Fujishiro T, Yamada M, Isobe S (2010) An interspecific linkage map of SSR and intronic polymorphism markers in tomato. Theor Appl Genet 121:731–739
Shultz JL, Kazi S, Bashir R, Afzal JA, Lightfoot DA (2007) The development of BAC-end sequence-based microsatellite markers and placement in the physical and genetic maps of soybean. Theor Appl Genet 114:1081–1090
Smulders MJ, Bredemeijer G, Rus-Kortekaas W, Arens P, Vosman B (1997) Use of short microsatellites from database sequences to generate polymorphisms among Lycopersicon esculentum cultivars and accessions of other Lycopersicon species. Theor Appl Genet 97:264–272
Stamova BS, Chetelat RT (2000) Inheritance and genetic mapping of cucumber mosaic virus resistance introgressed from Lycopersicon chilense into tomato. Theor Appl Genet 101:527–537
Suliman-Pollatschek S, Kashkush K, Shats H, Hillel J, Lavi U (2002) Generation and mapping of AFLP, SSRs and SNPs in Lycopersicon esculentum. Cell Mol Biol Lett 7:583–597
Tanksley SD, Ganal MW, Prince JP, De Vicente MC, Bonierbale MW, Broun P, Fulton TM, Giovannoni JJ, Grandillo S, Martin GB, Messeguer R, Miller JC, Miller L, Paterson AH, Pineda O, Roder MS, Wing RA, Wu W, Young ND (1992) High-density molecular linkage maps of the tomato and potato genomes. Genetics 132:1141–1160
Tautz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucl Acid Res 12:4127–4138
Thomas MR, Scott NS (1993) Microsatellite repeats in grapevine reveal DNA polymorphisms when analyzed as sequence-tagged sites (STSs). Theor Appl Genet 86:985–990
Thoquet P, Olivier J, Sperisen C, Rogowsky P, Laterrot H, Grimsley NH (1996) Quantitative trait loci determining resistance to bacterial wilt in tomato cultivar Hawaii7996. Mol Plant Microbe Interact 9:826–836
Trognitz BR, Schmiediche PE (1993) A new look at the incompatibility relationships in higher plants. Sex Plant Reprod 6:183–190
van Ooijen JW, Voorrips RE (2001) JoinMap® 3.0: Software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Voorrips RE (2002) Mapchart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang JF, Olivier J, Thoquet P, Mangin B, Sauviac L, Grimsley NH (2000) Resistance of tomato line Hawaii7996 to Ralstonia solanacearum Pss4 in Taiwan is controlled mainly by a major strain-specific locus. Mol Plant Microbe Interact 13:6–13
Yaghoobi J, Yates JL, Williamson VM (2005) Fine mapping of the nematode resistance gene Mi-3 in Solanum peruvianum and construction of a S. lycopersicum DNA contig spanning the locus. Mol Gen Genomics 274:60–69
Yi G, Lee JM, Lee S, Choi D, Kim BD (2006) Exploitation of pepper EST-SSRs and an SSR-based linkage map. Theor Appl Genet 114:113–130
Zhang LP, Khan A, Niño-Liu D, Foolad MR (2002) A molecular linkage map of tomato displaying chromosomal locations of resistance gene analogs based on a Lycopersicon esculentum × Lycopersicon hirsutum cross. Genome 45(1):133–146
Acknowledgments
The authors would like to thank Dr. Peter Hanson for providing information on tomato accessions, and Ms. Chiou-fen Hsu for technical assistance. This study is supported by funding provided by GTZ 81070160: Deutsche Gesellshaft fuer Technishe Zusammenarbeit GmbH of Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geethanjali, S., Kadirvel, P., de la Peña, R. et al. Development of tomato SSR markers from anchored BAC clones of chromosome 12 and their application for genetic diversity analysis and linkage mapping. Euphytica 178, 283–295 (2011). https://doi.org/10.1007/s10681-010-0331-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-010-0331-8