
Abstract
The inheritance of two flowering traits of chrysanthemum, initial blooming time and the duration of flowering, was investigated using segregation within an F 1 population derived from a cross between the autumn-flowering ‘Yuhualuoying’ and the summer-flowering ‘Aoyunhanxiao’ cultivars. The analysis, based on a single segregating generation and the major gene plus polygene mixed inheritance model, showed that the inheritance of both traits was compatible with the presence of two pairs of major genes displaying additivity–dominance–epistasis, with additivity predominating. As the heritability of both pairs of major genes was high (initial blooming time ~65%, duration of flowering ~72%), it should be possible to select for both traits in early breeding generations. A marker-trait association analysis based on sequence-related amplified polymorphism (SRAP) genotyping uncovered 10 (initial blooming time) and 12 (duration of flowering) markers significantly associated with phenotype, cumulatively explaining, respectively, 46 and 54% of the variation. Some potentially useful markers were identified.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chrysanthemum, Chrysanthemum morifolium (Ramat.) Kitam, is one of the most popular ornamentals in the world. Its diversity with respect to growth habit and both inflorescence form and colour have ensured its economic importance. The demand for chrysanthemum production, encompassing cut-flower, garden, potted plants and ground-cover types, is increasing worldwide. The flowering traits of a variety determine its adaptation and productivity in a given agro-ecological zone (Hao et al. 2008a). In China, most cultivars flower in autumn, with only a few flowering in early summer. Therefore, protected cultivation has been adopted for year-round production of chrysanthemum, which is rather costly and laborious. Understanding the inheritance pattern of a target trait is necessary for its improvement. Most genetic studies in chrysanthemum have targeted the ornamental traits of inflorescence such as colour (Li et al. 2005) and floret form (Chen et al. 1991, 2003; Zhang et al. 2008, 2010a), as well as vegetative traits such as plant type (Jiang et al. 2003; Zhang et al. 2010a; Zhao et al. 2009a). Flowering is photoperiod, temperature and vernalization dependent, so its genetic control is expected to be complex. Flowering time is a quantitative trait in kiwifruit (Zhu et al. 2002), snapdragon (Martin and Stimart 2003, 2005), Vernonia galamensis (Baye and Becker 2005) and cotton (Anbessa et al. 2006; Hao et al. 2008a). Little is known regarding the genetic control of flowering in chrysanthemum, which has hindered the improvement of this trait to some extent.
Historically, quantitative traits have been analyzed by calculating the total genetic effect based on population means and genetic variance (Cheng et al. 2006; Rumpunen and Kviklys 2003), since a quantitative trait is generally believed to be controlled by many polygenes. The effects of genes controlling quantitative traits can vary in magnitude, with some showing strong genetic effects (major genes) and some showing only weak ones (minor genes) (Chen et al. 2008; Oyant et al. 2008; Thumma et al. 2010). Therefore, the inheritance of a quantitative trait may be determined by a few major genes as well as by many polygenes. Based on this idea, Gai et al. (2003) have developed the concept of a major gene plus polygene mixed inheritance model (mixed model) to analyze quantitative traits in plants. This requires a single segregating generation and several jointly segregating families (Wang and Gai 1997; Zhang et al. 2000), and has been successfully applied to the genetic analysis of a number of agronomically important quantitative traits (Chen et al. 2009; Guo et al. 2006; Hao et al. 2008b; Zeng and Pu 2006; Zhang et al. 2006). Quantitative trait locus (QTL) analysis has been attempted in the ornamental species lily (Abe et al. 2002), rose (Dugo et al. 2005; Oyant et al. 2008) and carnation (Yagi et al. 2006), and genetic linkage maps of chrysanthemum have recently been constructed (Zhang et al. 2010b). Among the major methods of QTL analysis, marker-based variance analysis has become the preferred means where only limited genetic linkage map information is available—such as in the tree species poplar (Huang et al. 2004a, b; Su et al. 2000), Taxus yunnanensis (Su et al. 2009) and Camellia sinensis (Yao et al. 2010).
The sequence-related amplified polymorphism (SRAP) technique amplifies open reading frames. It has been used to understand genetic diversity (Budak et al. 2004; Hao et al. 2008b), construct genetic (Li and Quiros 2001) and transcriptome (Li et al. 2003) maps, and for gene tagging (Li et al. 2003; Han et al. 2008; Pan et al. 2003). Recently, we have reported the use of SRAP markers in chrysanthemum (Zhang et al. 2009, 2010b). Here, we describe a genetic analysis of initial blooming time and the duration of flowering in chrysanthemum, based on SRAP genotyping. We believe that these data will contribute to an improved understanding of the inheritance of these two flowering traits, and perhaps lay the foundations for future gene cloning and marker-assisted breeding in chrysanthemum.
Materials and method
Materials
The female mapping parent ‘Yuhualuoying’ flowers in autumn, while the male ‘Aoyunhanxiao’ flowers in summer. Both varieties were bred by the Chrysanthemum Germplasm Resource Preserving Centre, Nanjing Agricultural University, China. Artificial hybridization was used to make the ‘Yuhualuoying’ × ‘Aoyunhanxiao’ cross to generate a population of 142 F 1 progeny (Liu et al. 2010; Sun et al. 2010).
Design of field experiments and investigation of flowering traits
The field experiments were carried out in the nursery of the Chrysanthemum Germplasm Resource Preserving Centre in 2008 and 2009. The 142 F 1 seedlings, along with both parental lines, were vegetatively propagated in April of both 2008 and 2009, and a month later, rooted cuttings were transplanted to a nursery field in a randomised block design with three replications. The size of the plot was 1.0 m × 60 m and the plant spacing was 35 cm × 35 cm. Field management followed standard commercial practices. Initial blooming time and the duration of flowering were measured in both years. The former was defined as the number of days between transplanting to the date on which approximately 50% of the total flower buds were half-opened and fully pigmented. The latter was defined as the number of days between initial blooming and the date on which approximately 10% of the total flowers appeared wilted.
Genomic DNA isolation and SRAP marker analysis
Total genomic DNA was extracted from young unexpanded leaves using CTAB procedure (Murray and Thompson 1980). A set of 30 SRAP primers (10 forward and 20 reverse) was designed according to Li and Quiros (2001), producing 200 primer combinations as a means of finding polymorphism between the parental cultivars. Informative primer combinations were used to genotype the mapping population. All PCR and electrophoretic procedures followed Zhang et al. (2010b).
Heterosis and genetic analyses
Heterosis was measured both by mid-parent heterosis (H m) and the ratio of mid-parent heterosis (RHm) (Li and Wu 1997). H m was defined as the difference between F m (the mean trait value of the F 1 progeny) and MPV (the mean trait value of the two parental lines). RHm was given by (F m − MPV)/MPV × 100%. Statistical analyses of the flowering traits and one-sample t-tests were performed using SPSS v11.5 software. Since both phenotypic and genetic data were consistent over the 2 years (not shown), the 2 year mean was used for trait values. A single segregating generation and the mixed inheritance model analysis method described by Gai et al. (2003) were used. Briefly, suppose under the modifications of polygene and environment, the effects of major gene in segregation generation display an independent normal distribution, then the whole segregation generation could be seen as a mixed distribution made up by many independent normal distributions. The single generation segregation analysis may involve the two genetic model of null or one pair of major genes (A model) and two pairs of major genes (B model) in 11 kinds with special reference to major gene. Statistical deduction can be regarded as an estimation of probability distribution of observed value according to the principle of entropy maximization suggested by Akaike (1977). Therefore, comparison of the goodness-of-fit of the practical frequency distributions with the standard curves can approximate the inheritance model of a quantitative trait. The most-fitting genetic model was chosen according to Akaike’s Information Criterion (AIC). The smallest AIC value is the most-fitting genetic model. Akaike (1977) suggested that the hypothesis maximizing the expected entropy should be selected as the most fitting model. For this purpose, based on good-of-fit and parsimony, the hypothesis that leads to the smallest AIC will be chosen. A calculated software was provided by Dr. Gai, National Centre for Soybean Improvement, State Key Laboratory of Crop Genetics and Germplasm Enhancement of Nanjing Agricultural University. Genetic parameters were estimated with the method as detailed in Wang and Gai (1997) and Gai et al. (2003).
In a mixed inheritance model, the phenotypic value (p) can be expressed as the sum of the population mean (m), major gene effects (g), polygene effects (c) and the environmental effect (e), i.e. p = m + g + c + e, where g is dependent on major gene effects, and both c and e are normally distributed. The heritability of each major gene h 2 mg was given by σ 2 mg /σ 2 p × 100%. Because this study was undertaken using a single segregating generation, neither the polygene nor the environmental variances could be estimated.
Marker-trait association analysis
Marker profiles were scored by assigning ‘0’ for the absence and ‘1’ for the presence of each polymorphic fragment. Only reproducible and well-defined bands were scored. Each marker was identified by the primer pair used and a suffix was attached to the designation, where multiple fragments were identified from a single primer combination. According to the marker-based method of Singh et al. (1991), for each marker, the segregating progeny were assigned into groups, formed by those scored as ‘0’ and those as ‘1’, and a one-way variance analysis was then performed for each marker in turn to detect any SRAP loci linked to the trait.
Results
Trait distribution and heterotic performance
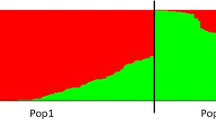
Initial blooming time and the duration of flowering segregated widely, with coefficients of variation of >9 and >21%, respectively. The skewness and kurtosis values (Table 1) and the frequency distributions (Fig. 1) indicated that both initial blooming time and the duration of flowering in the F 1 population fitted a normal distribution. There were highly significant differences between MPV and H m (Table 1). The RHm for initial blooming time was >12% and that for the duration of flowering ~6%, indicating a substantial level of heterosis for both traits. Transgressive segregation was present for both traits (Fig. 1), but transgressive heterosis was not observed (Table 1).

Frequency distributions for initial blooming time and the duration of flowering in an F 1 population derived from the cross between ‘Yuhualuoying’ (P 1) × ‘Aoyunhanxiao’ (P 2)
Goodness-of-fit tests
The goodness-of-fit of the 11 genetic models with the observed phenotypic data is reported in Table 2. Following the assumption that the model with the smallest AIC value is the best model, several models with the smallest AIC values were chosen for each trait. For initial blooming time, models B-1 and B-6 had the lowest AIC values, while for the duration of flowering the optimal models were A-1, A-4 and B-1. These models were therefore taken forward for further goodness-of-fit tests. Three tests for goodness-of-fit were then applied: equal distribution (U 21 , U 22 and U 23 ), Smirnov ( n W 2) and Kolmogorov test (D n ) (Table 3). According to Wang and Gai (1997), if the test statistics of all the models tested show significant differences, the model with the least significant statistic should be chosen, but if there are no significant differences, then the model with the smallest AIC should be preferred. On this basis, B-1 was selected for both traits.
Estimation of genetic parameters
Genetic parameters for initial blooming time and the duration of flowering were estimated using model B-1 (Table 4). For initial blooming time, the additive effect values of the two pairs of major genes were, respectively, 11.6 and 11.1, and their dominance effects 11.0 and 10.7. Thus, both the additive and dominance effects of the first pair of major genes were larger than those of the second pair. For the duration of flowering, the additive effect effects of the two pairs of major genes were 2.9 and 2.8, and their dominance effects were −2.8 and −0.5; once again, the additive and dominant effects of the first pair of major genes were larger than those of the second pair. The additive effects of the two pairs of major genes for initial blooming time and duration of flowering were larger that their dominance effects, indicating that the additive effects predominated. However, the size of the dominance effects of the two pairs of major genes was similar to that of the additive effects, suggesting that the two pairs of major genes showed over-dominance for both traits. The additivity × additivity, additivity × dominance, dominance × additivity and dominance × dominance interaction effects were all substantial (Table 4), indicating a significant level of epistasis between the two pairs of major genes. The heritability of the major genes for initial blooming time and duration of flowering were ~65 and 72%, respectively (Table 4).
SRAP marker analysis
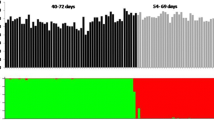
Of the 200 SRAP primer pairs screened against DNA of the two parental cultivars and 10 of the F 1 progeny, 62 were applied to the whole F 1 population, leading to the identification of 346 polymorphic fragments (5.6 per primer pair). The parental and a sample of 45 progeny profiles amplified with the Me13Em4 primer pair are shown in Fig. 2. The SRAP markers significantly associated with the flowering traits are listed in Table 5. Ten SRAP markers were significantly associated with initial blooming time and 12 with the duration of flowering. Both had a high cumulative contribution ratio of 45.54% for initial blooming time and 53.48% for duration of flowering. The contribution of any single marker to the phenotypic variance ranged from 3.0 to 10.1%.

Amplification profiles of chrysanthemum ‘Aoyunhanxiao’, ‘Yuhualuoying’ and their F 1 progeny with SRAP primer combination Me13Em4. M DNA marker, Y chrysanthemum ‘Yuhualuoying’, A chrysanthemum ‘Aoyunhanxiao’, 1–45 45 individuals of F 1 progeny, sequence of Me13 is 5′-TGAGTCCAAACCGGTAA-3′ and that of Em4 is 5′-GACTGCGTACGAATTTGA-3′
Discussion
Heterotic performance for two flowering traits
Heterosis is common in chrysanthemum. Chen et al. (2003) claimed that hybrid vigor of flower diameter, number of ray and tubular florets increased significantly in F 1 progeny of chrysanthemum of small inflorescences, and the increasing extend in F 1 population became slow with increase in flower diameter, number of ray and tubular florets of parental lines. The heterosis mainly originated from the heterogeneity between parental lines. In the current study, we have analysed a cross between an autumn- and a summer-flowering chrysanthemum. In the progeny population, both initial blooming time and the duration of flowering varied considerably. Heterosis was present for both traits, with evidence for over-dominance. Therefore, chrysanthemum breeding should greatly benefit from the heterosis available in wide crosses.
The genetics of flowering traits with reference to major genes
Chrysanthemum is largely self-incompatible and suffers from inbreeding depression (Anderson and Ascher 2000; Li and Chen 2007; Xu et al. 2009). This makes it practically difficult to obtain advanced generations of inbred lines of the sort used for genetic research in self-compatible species. A number of tree species are similarly heterozygous, and in these the F 1 generation is generally used as a pseudo-F 2 population for genetic studies (Grattapaglia and Sederoff 1994; Marron and Ceulemans 2006; Song et al. 2005; Wang et al. 2009). Such a pseudo-F 2 population was also widely applied in horticultural crops of self-incompatibility such as artichoke (Lanteri et al. 2006; Portis et al. 2009) and garlic (Ma et al. 2006). In chrysanthemum, flower diameter, central flower diameter, and ray and tubular floret number are all predominantly controlled by additive genes (Ghimiray et al. 2005; Pal and George 2002; Sirohi and Behera 2000; Zhang et al. 2008, 2010a), but little is known of the inheritance of flowering traits. Although flowering time depends on planting date, photoperiod, temperature and other climatic factors (Chardon et al. 2004), genes also play an important part (Anbessa et al. 2006; Cai et al. 2008; Godoy and Palomo 1999; Hao et al. 2008a). The outcome of the present genetic analysis was that both flowering traits were compatible with the action of two pairs of major genes displaying additivity–dominance–epistasis. Their contribution to the phenotypic variation was substantial and their heritabilities were as high as 0.88. Thus, it should be relatively easy to select for these two traits in the early generations of a breeding programme. The additive effect of these major genes predominated, although both dominance and epistasis were detected. Theoretically, both dominance and epistasis in heterosis could be adopted in a breeding programme. Although we were unable to estimate the contribution of polygene and environmental variation, as is possible using several jointly segregating families (Chen et al. 2009; Zeng and Pu 2006), the detection of major genes for initial blooming time and duration of flowering of chrysanthemum is a useful start to identifying molecular markers associated with these traits.
SRAP markers associated with flowering traits of chrysanthemum
Genetic linkage maps, molecular markers and QTL analysis have been attempted in a number of ornamental crops (Abe et al. 2002; Dugo et al. 2005; Dunemann et al. 1999; Remay et al. 2009; Yagi et al. 2006). In chrysanthemum, in contrast, there has been less progress to date in this direction (Zhang et al. 2010b; Zhao et al. 2009a, b). Here, we have identified SRAP markers associated with initial blooming time and the duration of flowering were identified by the marker-based variance method based on 2-year phenotypic data in an F 1 (pseudo-F 2) population. That the contribution of each individual marker explained 3–10% suggested that the genetic effects of quantitative trait genes controlling flowering traits of chrysanthemum varied in magnitude, which further confirmed the detection of major genes for the flowering traits described above. Moreover, some significant differences at the 0.05 or 0.01 level were detected between every two significantly associated markers by double-factor variance analysis for initial blooming time and duration of flowering, respectively (data not shown), which also provides some molecular evidence for the above-mentioned interaction of the two pairs of major genes.
Compared to marker-trait analyses performed in other crops (Huang et al. 2004a; Li and Zheng 1998; Su et al. 2009), the contribution of a single marker locus to the phenotypic variation of flowering traits studied here is relatively low. However, we believe that some useful SRAP markers of flowering behaviour QTL have been detected in this study. For example, the locus Me20Em15-1 explained ~8.5% of the variation in initial blooming time and approximately 4.1% in the duration of flowering, as did Me13Em4-8 and Me20Em1-1. As SRAP marker amplifies open reading frames, therefore, the associated genetic markers with relatively high contributions to the phenotypic variation of flowering traits identified here may be useful in target gene cloning.
The marker-based variance method is limited, especially as it has difficulty in determining whether the marker loci are associated with one or more QTL, and cannot easily define intra-chromosomal QTL location with any precision. It also tends to underestimate the size of the genetic effect, and suffers from a high false positive rate in the detection of genetic markers or QTL associated with a trait (Huang et al. 2004b; Tanksley et al. 1982). The outcome of marker-based variance, however, is generally quite consistent with map-based methods in terms of approximate QTL location (Huang et al. 2004b; Li et al. 1999; Su et al. 2000). Thus, the SRAP marker analysis will help to understand the inheritance pattern of flowering traits of chrysanthemum. Even so, further research using map-based QTL analysis is needed to accurately map horticulturally important traits like the flowering behaviour of chrysanthemum.
References
Abe H, Nakano M, Nakatsuka A, Nakayama M, Koshioka M, Yamagishi M (2002) Genetic analysis of floral anthocyanin pigmentation traits in Asiatic hybrid lily using molecular linkage maps. Theor Appl Genet 105:1175–1182
Akaike H (1977) On entropy maximisation principle. In: Krishnaiah PR (ed) Proceedings of the symposium on applications of statistics, Amsterdam, The Netherlands, pp 27–47
Anbessa Y, Warkentin T, Vanderberg A, Ball R (2006) Inheritance of time to flowering in chickpea in a short-season temperate environment. J Hered 97:55–61
Anderson NO, Ascher PD (2000) Fertility changes in inbred families of self-incompatible chrysanthemums (Dendranthema grandiflorum). J Am Soc Hortic Sci 125(5):619–625
Baye T, Becker HC (2005) Genetic variability and interrelationship of traits in the industrial oil crop Vernonia galamensis. Euphytica 142:119–129
Budak H, Shearman RC, Parmaksiz I (2004) Molecular characterization of buffalograss germplasm using sequence-related amplified polymorphism markers. Theor Appl Genet 108:328–334
Cai CC, Tu JX, Fu TD, Chen BY (2008) The genetic basis of flowering time and photoperiod sensitivity in rapseed Brassica napus L. Russ J Genet 44(3):326–333
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A (2004) Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168:2169–2185
Chen YZ, Jin BM, Wu SF, He XD, Yao GS (1991) The performance of some traits of F1 generation of chrysanthemum varietal hybridization. Acta Hortic Sin 18(3):258–262
Chen FD, Jiang JF, Guo WM (2003) Heredity of several flower characters in Dendranthema grandiflorum with small inflorescences. Acta Hortic Sin 30(2):175–182
Chen CX, Cai SB, Bai GH (2008) A major QTL controlling seed dormancy and pre-harvest sprouting resistance on chromosome 4A in a Chinese wheat landrace. Mol Breed 21(3):351–358
Chen SL, Li YR, Cheng ZS, Liao BS, Lei Y, Liu JS (2009) Heterosis and genetic analysis of oil content in peanut using mixed model of major gene and polygene. Sci Agric Sin 42(9):3048–3057
Cheng CH, Seal AG, Murphy SJ, Lowe GR (2006) Variability and inheritance of flowering time and duration in Actinidia chinensis (kiwifruit). Euphytica 147:395–402
Dugo ML, Satovic Z, Millán T, Cubero JI, Rubiales D, Cabrera A, Torres AM (2005) Genetic mapping of QTLs controlling horticultural traits in diploid roses. Theor Appl Genet 111:511–520
Dunemann F, Kahnau R, Stange I (1999) Analysis of complex leaf and flower characters in Rhododendron using a molecular linkage map. Theor Appl Genet 98:1146–1155
Gai JY, Zhang YM, Wang JK (2003) Genetic system of quantitative traits in plants. Science Press, Beijing, pp 96–102
Ghimiray TS, Indrajit S, Amar R (2005) Variability studies in chrysanthemum grown over two environments. Res Crops 6(3):514–516
Godoy AS, Palomo GA (1999) Genetic analysis of earliness in upland cotton (Gossypoum hirsutum L.). I. Morphological and phenological variables. Euphytica 105:155–160
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Guo ZG, Ding P, He LY, Xu CG (2006) Genetic analysis of agricultural traits in rice related to phosphorus efficiency. Acta Genet Sin 33(7):634–641
Han XY, Wang LS, Liu ZA, Jan DR, Shu QY (2008) Characterization of sequence-related amplified polymorphism markers analysis of tree peony bud sports. Sci Hortic 115:261–267
Hao JJ, Yu SX, Ma QX, Fan SL, Song MZ (2008a) Inheritance of time of flowering in upland cotton under natural conditions. Plant Breed 127:383–390
Hao Q, Liu ZA, Shu QY, Zhang RE, Jan DR, Wang LS (2008b) Studies on Paeonia cultivars and hybrids identification based on SRAP analysis. Hereditas 145:38–47
Huang LJ, Su XH, Zhang XH, Huang QJ (2004a) SSR molecular markers related to wood density and fibre traits in poplar. Acta Genet Sin 31(3):299–304
Huang QJ, Su XH, Huang LJ, Zhang ZY (2004b) QTLs mapping for wood properties of Populus deltoides × P. cathayana. Sci Silvae Sin 40(2):55–60
Jiang JF, Chen FD, Guo WM (2003) Heredity of several characters in Dendranthema grandiflorum with small inflorescences. J Nanjing Agric Univ 26(2):11–15
Lanteri S, Acquadro A, Comino C, Mauro R, Mauromicale G, Portis E (2006) A first linkage map of globe artichoke (Cynara cardunculus var scolymus L.) based on AFLP, S-SAP, M-AFLP and microsatellite markers. Theor Appl Genet 112:1532–1542
Li XL, Chen FD (2007) Self-incompatibility in Dendranthema morifolium. J Wuhan Bot Res 25(6):591–595
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Li B, Wu R (1997) Heterosis and genotype × environment interactions of juvenile aspens in two contrasting sites. Can J For Res 27(10):1525–1537
Li CL, Zheng KL (1998) RAPD-based analysis for the QTLs related to plant height and heading date. Acta Genet Sin 25(1):34–39
Li JH, Su XH, Zhang QW, Louis Z (1999) Detection of QTLs for growth and phenology traits of poplar using RAPD markers. For Res 12(2):111–117
Li G, Gao M, Yang B, Quiros CF (2003) Gene for gene alignment between the Brassica and Arabidopsis genomes by direct transcriptome mapping. Theor Appl Genet 107:168–180
Li MT, Yu LJ, Wang LM, Liu JM, Lei C (2005) The heredity of flower colors and the discovery of flower color chimera in chrysanthemum species. Hereditas (Beijing) 27(6):948–952
Liu SY, Zhang F, Chen SM, Chen FD (2010) Interspecific hybridization between the tetraploid Chrysanthemum nakingense and Ch. grandiflorum ‘Zhongshanzixing’ and the genetic performance of their F1 hybrids. Sci Agric Sin 43(12):2500–2507
Ma SB, Guo RL, Nie YX, Nie HQ (2006) Combining ability and genetic analysis of yield characters for fragrant-flowered garlic. Acta Hortic Sin 33(1):78–83
Marron N, Ceulemans R (2006) Genetic variation of leaf traits related to productivity in a Populus deltoides × P. nigra family. Can J For Res 36:340–390
Martin WJ, Stimart DP (2003) Early generation evaluation in Antirrhinum majus for prediction of cutflower postharvest longevity. J Am Soc Hortic Sci 128:876–880
Martin WJ, Stimart DP (2005) Genetic analysis of advanced populations in Anterrhinum majus L. with special reference to cut flower postharvest longevity. J Am Soc Hortic Sci 130:434–441
Murray M, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Oyant LHS, Crespel L, Rajapakse S, Zhang L, Foucher F (2008) Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet Genomes 4:11–23
Pal P, George SV (2002) Genetic variability and correlation studies in chrysanthemum. Hortic J 15(2):75–81
Pan QH, Hu ZD, Tanisaka T, Wang L (2003) Fine mapping of the blast resistance gene Pil5, linked to Pii on rice chromosome 9. Acta Bot Sin 45:871–877
Portis E, Mauromicale G, Mauro R, Acquadro A, Scaglione D, Lanteri S (2009) Construction of a reference molecular linkage map of globe artichoke (Cynara cardunculus var. scolymus). Theor Appl Genet 120:59–70
Remay A, Balanne D, Thouroude T, Couviour FL, Oyant LHS, Foucher F (2009) A survey of flowering genes reveals the role of gibberellins in floral control in rose. Theor Appl Genet 119:767–781
Rumpunen K, Kviklys D (2003) Combining ability and patterns of inheritance for plant and fruit traits in Japanese quince (Chaenomeles japonica). Euphytica 132:139–149
Singh PS, Gutierrez JA, Molina A, Urrea C, Gepts P (1991) Genetic diversity in cultivated common bean: II Marker-base analysis of morphological and agronomic traits. Crop Sci 31:24–39
Sirohi PS, Behera TK (2000) Genetic variability in chrysanthemum. J Ornam Hortic (New Series) 3(1):34–36
Song RG, Lu WP, Li CY, Lin XG, Shen YJ, Li XH (2005) Heridity analysis of fruit mature period in progenies from interspecific and intraspecific hybridization of Vitis amurensis Rupr. Acta Hortic Sin 32(2):212–217
Su XH, Li JH, Chen BW, Zhang QW, Zhang XH (2000) Detection and identification of molecular markers associated with quantitative traits of leaf in poplar. Sci Silvae Sin 36(1):34–36
Su JR, Miao YC, Zhang ZJ (2009) RAPD markers related to taxol content of Taxus yunnanensis. Sci Silvae Sin 45(7):16–20
Sun CQ, Chen FD, Teng NJ, Liu ZL, Fang WM, Hou XL (2010) Factors affecting seed set in the crosses between Dendranthema grandiflorum and its wild species. Euphytica 191:181–192
Tanksley SD, Medina-Hilho H, Rick CM (1982) Use of naturally-occurring enzyme variation to detect and map gene controlling quantitative traits in an interspecific backcross of tomato. Heredity 49:11–25
Thumma BR, Southerton SG, Bell JC, Oven JV, Henery ML, Moran GF (2010) Quantitative traits locus (QTL) analysis of wood quality traits in Eucalyptus nitens. Tree Genet Genomes 6:305–317
Wang JK, Gai JY (1997) Identification of major gene and polygene mixed inheritance model and estimation of genetic parameters of a quantitative trait from F2 progeny. Acta Genet Sin 24(5):432–440
Wang X, Wadl PA, Rinehart TA, Scheffler BE, Windham MT, Spiers JM, Johnson DH, Trigiano RN (2009) A linkage map for flowering dogwood (Cornus florida L.) based on microsatellite markers. Euphytica 165:165–175
Xu YF, Chen FD, Teng NJ, Chen SM, Li FT (2009) Preliminary study on inbreeding depression of Dendranthema morifolium. J Plant Res Environ 18(4):28–32
Yagi M, Onosaki T, Taneya M, Watanabe H, Yoshimura T, Yoshinari T, Ochiai Y, Shibata M (2006) Construction of a genetic linkage map fro the carnation by using RAPD and SSR markers and mapping quantitative trait loci (QTL) for resistance to bacterial wilt caused by Burkholderia caryophylli. J Jpn Soc Hortic Sci 75(2):166–172
Yao MZ, Qiao TT, Ma CL, Jin JQ, Chen L (2010) The association analysis of phenotypic traits with EST-SSR markers in tea plants. J Tea Sci 30(1):45–51
Zeng YW, Pu XY (2006) Genetic analysis for cold tolerance at booting stage for japonica rice (Oryza sativa L.). Indian J Genet Plant Breed 66(2):100–102
Zhang YM, Gai JY, Zhang MC (2000) Jointly segregating analysis of P1 P2 F1 and F2 or F2:3 families. J Southwest Agric Univ 22(1):6–9
Zhang SF, Ma CZ, Zhu JC, Wang JP, Wen YC, Fu TD (2006) Genetic analysis of oil content in Brassica napus L. using mixed model of major gene and polygene. Acta Genet Sin 33(2):171–180
Zhang F, Fang WM, Chen FD, Zhao HB, Jia WK (2008) Genetic variability patterns and correlation analysis for cut-chrysanthemum with emphasis on inflorescence traits. J Zhejiang For Coll 25(3):293–297
Zhang F, Chen FD, Fang WM, Li FT, Liu PS (2009) Optimization and establishment of SRAP-PCR reaction system of Dendranthema × grandiflorum. J Plant Res Environ 18(3):44–49
Zhang F, Fang WM, Chen FD, Chen SM (2010a) Combining ability analysis on ornamental characters of chrysanthemum. Acta Hortic Sin 37(4):589–596
Zhang F, Chen SM, Chen FD, Fang WM, Li FT (2010b) SRAP-based mapping and QTL detection for inflorescence-related traits in chrysanthemum (Dendranthema morifolium). Mol Breed. doi:10.1007/s11032-010-9409-1
Zhao JY, Chen FD, Teng NJ, Chen SM (2009a) Genetic analysis and RAPD marker of creeping habits in ground-cover chrysanthemum. Sci Agric Sin 42(2):734–741
Zhao JY, Chen SM, Chen FD (2009b) Conversion of RAPD marker linked to creep plant type in ground-cover chrysanthemum to SCAR marker. Sci Silvae Sin 45(9):147–150
Zhu D, Lawes GS, Gordon IL (2002) Estimates of genetic variability for vegetative and reproductive characters of kiwifruit (Actinidia deliciosa). Euphytica 124:93–98
Acknowledgments
This project was supported by the National Nature Science Foundation of China (Grant No. 30871724), Program for New Century Excellent Talents in University of Chinese Ministry of Education (Grant No. NCET-06-0489), and 948 Project of Ministry of Agriculture (Grant No. 2008-G3).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, F., Chen, S., Chen, F. et al. Genetic analysis and associated SRAP markers for flowering traits of chrysanthemum (Chrysanthemum morifolium). Euphytica 177, 15–24 (2011). https://doi.org/10.1007/s10681-010-0239-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-010-0239-3